 2017, Vol. 37
2017, Vol. 37



生物炭是生物质废弃物在限氧热解( < 700 ℃)条件下产生的一类高度芳香化富碳物质(Li et al., 2011), 主要由C、H、O等元素组成, 微观上为紧密堆积的芳香环片, 含有—COOH、—OH等官能团.生物炭具有明显的多孔特征, 且具有较大的比表面积与表面能(Cornelissen et al., 2005), 有研究发现, 生物炭表面具有较大的负电荷与电荷密度(Liang et al., 2006).生物炭具有高度的稳定性, 施入土壤后不易被微生物降解, Kuzyakov等(2014)利用了14C标记法分析了生物炭的滞留时间, 结果显示, 在理想条件下生物炭的平均滞留时间可以达到2000年.还有研究表明, 与生物质直接施入土壤被分解产生CO2相比, 制备成生物炭能够多留存至少40%的有机碳(Lehmann et al., 2010).从而证实了其作为长期稳定碳库的可能性, 因此, 生物炭具有影响全球热平衡和气候变化的重要意义(Schmidt et al., 2011).生物炭不仅可以起到长期固碳作用, 还能够改良土壤性质, 修复污染土壤, 这些性质都与生物炭施入土壤后的稳定性密切相关.
生物炭在进入土壤后会在干湿交替、灌溉翻耕等自然和人为条件下释放出可溶态生物炭, 游离的可溶态生物炭较不稳定, 易被微生物分解.进入土壤溶液的可溶态生物炭同时会与土壤各组分如腐殖质、微生物、土壤矿物质(Kalbitz et al., 2005)等发生反应, 这种反应可能会减少可溶态生物炭与微生物的接触, 从而避免其被微生物分解.土壤矿物质(如高岭土、针铁矿等)是土壤的重要组成成分, 它们对土壤自身有机质的吸附保护作用可以提高其自身有机质的稳定性(Kaiser et al., 2000).目前已有的研究主要聚焦于整体生物炭与矿物质的界面行为及其对于生物炭稳定性的影响(Nguyen et al., 2008; Sollins et al., 2009; Yang et al., 2016).然而, 可溶态生物炭与土壤矿物质结合对生物炭稳定性影响的研究鲜见报道.
本研究假设核桃壳生物炭中释放出的可溶态生物炭能够与土壤矿物质高岭土及针铁矿发生结合行为, 同时这种结合可以提高可溶态生物炭的稳定性.为了验证以上假设, 本研究开展批次吸附实验及微生物降解实验, 探讨高岭土、针铁矿对可溶态生物炭的结合机理, 考察与矿物质结合后的可溶态生物炭的微生物降解性.
2 材料与方法(Materials and methods) 2.1 材料与试剂在本实验中, 选取核桃壳作为原材料, 因为核桃壳生物炭的无机矿物组分含量较少(Zhao et al., 2013), 可以最大程度避免来自生物炭自身矿物组分对于实验的影响.选取的粘土矿物质为高岭土(Al2O3·2SiO2·2H2O)和针铁矿(α-FeO(OH)), NaCl、NaH2PO4、CaCl2、(NH4)2SO4和KH2PO4均为分析纯, 上述粘土矿物和药品均采购于国药集团化学试剂有限公司.
提取微生物接种液的土壤样本采自江苏省常熟市(中国科学院常熟农业生态实验站), 该市位于长三角地区, 太湖西岸, 属于亚热带暖湿季风气候.年平均气温15.7 ℃, 年平均降水量约为1130 mm.供试稻田土为潮土(潮湿新成土), 成土母质为湖相沉淀物.采样深度为土壤表层0~20 cm, 取回后风干磨碎, 全部过2 mm筛.土壤pH为6.31, DOC含量为1.00 g·kg-1, 碳含量为26.6 g·kg-1, 氮含量为1.81 g·kg-1, C/N比为14.7.
2.2 实验方法 2.2.1 可溶态生物炭的制备与表征将核桃壳在自然条件下风干破碎, 全部过2 mm筛.将经过预处理的核桃壳颗粒放在自制的生物炭热解反应器内(Zhao et al., 2013).充入N2以排出内部的O2, 然后将装置放在马弗炉内.本实验所使用的生物炭是在升温速率控制为20 ℃·min-1, 500 ℃下加热4 h制得的, 制得的生物炭研磨全部过1 mm筛.
将土壤浸提液过0.45 μm滤膜是区分含水土层中可溶态物质和不溶态颗粒物的常用方法(Amy et al., 1992; Aiken et al., 2011), 本实验参照此方法, 将能够通过0.45 μm滤膜的生物炭定义为可溶态生物炭.为了避免对后续可溶态生物炭与土壤矿物质的结合实验造成干扰, 本研究中的生物炭浸提液采用不添加背景电解质的去离子水.参照目前常用的可溶态生物炭制备方法(Qu et al., 2016), 具体过程如下:将30 g新鲜生物炭用3 L去离子水浸没, 25 ℃避光恒温条件下以200 r·min-1搅拌48 h, 溶液过0.45 μm滤膜, 滤液作为可溶态生物炭贮备液冷藏保存.可溶态生物炭的碳含量用TOC仪测量(TOC-VCPN, 日本岛津公司).通过气相色谱质谱联用仪(GCMS-QP2010, 日本岛津公司)检测可溶态生物炭组分.
2.2.2 生物炭的表征pH的测定:将0.125 g生物炭放入25 mL水中, 振荡1 h, 然后静置5 min, 用pH计(pH510型, Eutech Instruments Pte Ltd)测定pH.元素分析:利用灼烧法, 用元素分析仪测定固体生物炭中C、H、O、N的含量.比表面积的测定:采用BET-N2分析仪(ASAP 2010 M+C, Micromeritics Inc., USA)测定生物炭的比表面积.CEC的测定:利用BaSO4强迫交换法测定生物炭的阳离子交换量.
2.2.3 土壤矿物质吸附可溶态生物炭矿物质吸附可溶态生物炭的实验中, 分别用0.01 mol·L-1 CaCl2、0.01 mol·L-1 NaCl和0.01 mol·L-1 NaCl-NaH2PO4作为背景电解质液, 前两者背景溶液可直接将其化学物质溶于去离子水中制得; 对于NaCl-NaH2PO4, 在NaCl背景溶液的离心管中再加入1 mL 50 mmol·L-1的NaH2PO4溶液, 用0.1 mol·L-1 NaOH或者0.1 mol·L-1 HCl调节pH=4, 在每种背景溶液中加入可溶态生物炭, 使其浓度达到5、10、20、30、40、60、80、100 mg·L-1, 加入0.04 μmol·L-1 HgCl2抑制微生物生长, 溶液的pH值再调至4.
矿物质的预处理:去除高岭土和针铁矿中的碳酸盐和自身有机质.用1 mol·L-1的NaCl溶液(矿物质质量与NaCl溶液体积比为1:5(g/L))清洗高岭土使其中所有的金属离子全部转变成Na+形式, 冷冻干燥, 过200 μm筛(Mikutta et al., 2007), 测量矿物质的Zeta电位, 高岭土的Zeta电位为-28.1 mV, 针铁矿的Zeta电位为29.0 mV.
分别用0.01 mol·L-1 CaCl2、0.01 mol·L-1 NaCl和0.01 mol·L-1 NaCl-NaH2PO4溶液制备矿物质贮备液(20 g·L-1), 调节至pH=4.0.然后将5 mL的矿物质悬浮液加入到25 mL的可溶态生物炭溶液中, 最终溶液的pH值再调至4.在避光、95 r·min-1转速和(25±2) ℃条件下水平振荡24 h达到平衡, 然后离心, 上层清液通过0.45 μm滤膜.测量滤液中的可溶态生物炭浓度.吸附前和吸附平衡后的可溶态生物炭浓度差即为被吸附的可溶态生物炭, 固相冷冻干燥保存.
本实验中3种背景溶液分别代表 3种不同的作用力(Mikutta et al., 2007), CaCl2背景溶液中, 含有Ca2+离子架、配体交换和范德华力3种作用机制; NaCl中含有配体交换与范德华力两种作用机制; NaCl+NaH2PO4中加入的H2PO4-会附着在矿物质表面形成磷酸化的组织(Mikutta et al., 2006), 因此, 只有范德华力作用.可通过三者之间的吸附量的差值推断各自吸附机理所占总吸附量的比例, 不同机理的吸附量(Ca2+架桥、配体交换、范德华力)通过公式(1)~(3)计算.
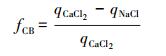
|
(1) |
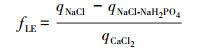
|
(2) |
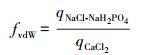
|
(3) |
式中, fCB、fLE、fvdW分别表示以Ca2+架桥、配体交换、范德华力为主导的吸附所占的比例, qCaCl2、qNaCl、qNaCl-NaH2PO4分别代表CaCl2、NaCl、NaCl+NaH2PO4背景体系中的矿物质对于可溶态生物炭的吸附量.
2.2.4 微生物降解实验微生物接种液与营养液的制备:微生物接种液的制备参考文献方法(Zimmerman, 2010), 取上述稻田土, 按土水比为1:5(m/V, g/L)加去离子水, 曝气培养24 h, 离心后过0.45 μm滤膜.微生物营养液的制备参考文献(Li et al. 2014)方法, 称取60 g (NH4)2SO4和6 g KH2PO4溶解于去离子水并定容至1 L, 调节pH=4, 配置微生物营养液.
培养实验:称取2.2.3节步骤中吸附可溶态生物炭后冷冻干燥的干矿物质(选取可溶态生物炭初始浓度为80 mg·L-1的处理组)使干矿物质吸附的可溶态生物炭为0.05 g, 放置于玻璃瓶中, 加入20 mL营养液与0.4 mL接种液.对于每种矿物质, 共设置纯矿物质(作为空白对照以扣除土壤浸提液本身含有的有机质)、矿物质+CaCl2、矿物质+NaCl、矿物质+NaCl-NaH2PO4、纯可溶态生物炭5种处理.气相色谱仪测量瓶内CO2浓度, 称重法补水.采样时间点选取为1、3、7、14、21、28、42、56 d(Mikutta et al., 2007).
为了获得可溶态生物炭CO2的释放速率和最大释放量, 可溶态生物炭CO2累计释放曲线采用双指数模型拟合(Mikutta et al., 2007), 将可溶态生物炭分为稳定与不稳定两部分, 公式如下:
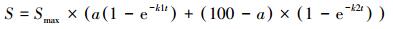
|
(4) |
式中, S为可溶态生物炭t时刻的矿化百分比; Smax为最大矿化百分比; a为可溶态生物炭可矿化部分中不稳定碳的比例; k1为可溶态生物炭中可矿化部分不稳定碳的一阶矿化速率常数(d-1), k2为可溶态生物炭中稳定碳的一阶矿化速率常数(d-1), t为培养时间(d)
3 结果与讨论(Results and discussion) 3.1 生物炭的表征由表 1可知, 以核桃壳为原料制成的生物炭的pH=8.22, 与大多数生物炭呈碱性的结论(Jones et al., 2011)一致.生物炭的主要成分是碳, 其含量高达88%.以往的研究表明, 生物炭主要是由碳元素组成的各种芳香族化合物的混合物(Goldberg, 1985; Glaser et al., 1998).生物炭除了具有丰富的碳外, 还含有23.3 g·kg-1灰分, 主要是由矿质元素如Ca、K、Si等组成(表 1).可溶态生物炭含量为10.3 g·kg-1, 其主要成分为小分子醇类(如1-乙氧基-2-丙醇等)、酯类(如邻苯二甲酸丁酯等), 以及少量醛酮与酚类, 它们的占比分别为32.4%、17.7%、5.0%、5.0%.
| 表 1 生物炭基本性质 Table 1 Basic properties of biochar |
土壤矿物质对有机质的吸附保护被认为是一种很重要的有机质稳定化过程(Kaiser et al., 2000).土壤矿物质吸附有机质可以减少其被微生物降解的速率, 这是因为有机质与矿物质之间存在化学或物理作用力(Kalbitz et al., 2005).矿物质吸附有机质的机理包括库仑力(氢键作用力、阳离子架桥、配体交换)与非库仑力(Arnarson et al., 2000; Feng et al., 2005).其中, 最主要的吸附机理为配体交换、阳离子架桥与范德华力.本实验中, 3种背景溶液分别代表一种或多种作用力, 根据图 1和公式(1)~(3)计算得到各自作用力所占总吸附量的比例.
 |
| 图 1 高岭土(a)与针铁矿(b)的吸附曲线 Fig. 1 Adsorption curve of kaolinite (a) and goethite (b) |
由高岭土的吸附曲线(图 1a)可以看出, 3种作用力随着可溶态生物炭初始浓度的增大而增大, 但它们的相对大小在不同浓度范围内不一致.在可溶态生物炭初始浓度较低时(如20 mg·L-1), Ca2+架桥占65.1%, 配体交换占30.3%, 范德华力占0.5%.而当初始浓度较高时(如80 mg·L-1), Ca2+架桥占21.4%, 配体交换占2.7%, 范德华力占75.9%.说明在浓度较低时, 结合力较强的Ca2+架桥和配体交换为主导, 当浓度增大时, Ca2+架桥和配体交换受到吸附位点的限制, 范德华力虽然结合力较弱, 但不受位点限制, 故当浓度增大时, 成为主导的吸附机理.在一些研究中发现, Ca2+架桥的吸附力>配体交换>范德华力(Mikutta et al., 2007), 这与本实验的结论相符合.
由图 1b可知, 针铁矿的吸附量大于高岭土, 这与针铁矿呈正电性(Zeta电位29.0 mV)而高岭土呈负电性(Zeta电位-28.1 mV)有关.对于吸附带负电荷的生物炭(Bell et al., 2011), 针铁矿的静电吸附更明显, 并且与高岭土明显不同, 针铁矿吸附的3种作用力随着可溶态生物炭初始浓度的增大先升高而后下降, 而且主要是范德华力, 少量表现出配体交换, Ca2+的存在不仅没有离子架桥作用, 相反抑制了其它两种作用力.
针铁矿吸附曲线的特殊性可能与其自身性质有关, 针铁矿(α-FeOOH)是热力学最稳定的铁氧化物, 具有较大比表面积(18~132 m2·g-1)(Strauss et al., 1997).针铁矿的层间结构能够被水化, 一些研究中指出, 针铁矿的水化结构中可能存在着在(H2O)-(H2O)-OH2-OH-Fe-O-O-Fe-R结构, 这表明存在两种不同结合水, 或双层水层结构(Ghose et al., 2010).研究表明, 外层水的结构更为活泼, 可能成为主要的吸附位点, 但这也导致了其较易被破坏(Catalano et al., 2008).当可溶态生物炭初始浓度上升时, 针铁矿的水化层结构被破坏, 吸附位点与比表面积减少, 从而导致吸附曲线下降.此外, 当可溶态生物炭初始浓度上升时, 部分吸附位点会因为结构内部的孔被大分子的可溶态生物炭堵塞而降低, 相关文献也报道了有机质吸附在矿物质表面而导致其吸附位点减少(Kaiser et al., 2003).此外, 有机质的存在也会使得针铁矿的吸附曲线呈现峰形即低浓度上升高浓度下降, 这可能是由于高浓度的有机质改变了针铁矿的表面电荷, 从而影响了针铁矿的吸附能力(左继超等, 2017).
在实验中, 加入的Ca2+可能与针铁矿及可溶态生物炭发生了反应, 推测其内在机理可能有以下几种: ①Ca2+与针铁矿中的Fe3+竞争吸附; ②Ca2+或者Fe3+与可溶态生物炭形成了弱吸附或难吸附的物质(Ali et al., 1996); ③在矿物质表面形成复杂的物质(Spark et al., 1997), 如矿物质-金属离子-可溶态生物炭复合物; ④Ca2+改变了矿物质中Fe的d电子云排布从而影响了矿物质的界面性质(Weng et al., 2005); ⑤生物炭呈负电性(Bell et al., 2011), 针铁矿Zeta电位呈正电性, 故Ca2+的加入会影响生物炭的负电性, 从而抑制了针铁矿与可溶态生物炭的静电吸引.这些原因可能导致加入Ca2+后, 吸附量下降.在真实土壤环境中, 结合机理更为复杂, 矿物质不同的结构和位置都会导致吸附机理的不同, 离子强度和pH也会影响吸附机理(Au et al., 1999; Namjesnik-Dejanovic et al., 2001).
3.3 微生物降解实验图 2是2种矿物质通过不同过程吸附可溶态生物炭后的微生物降解动力学曲线.将微生物降解可溶态生物炭的碳累计排放率用双指数模型拟合(Kalbitz et al., 2005), 利用得到的数据(表 2)进行计算得到半衰期.
 |
| 图 2 微生物降解被矿物质吸附可溶态生物炭的碳累积释放率(a.高岭土; b.针铁矿) Fig. 2 Accumulated C release rate of free dissolvable biochar and dissolvable biochar adsorbed by mineral (a. kaolinite, b. goethite) |
| 表 2 微生物降解双指数模型拟合数据 Table 2 Fitting of the microbial degradation by double exponential model |
如图 2所示, 可溶态生物炭在第1周被快速降解, 1周后, 降解速率变小, 6周后基本没有CO2释放出.经过8周培养, 可溶态生物炭大约有9%的碳以CO2的形式排放, 这说明可溶态生物炭也具有一定的稳定性.可溶态生物炭被高岭土吸附后, 抗微生物降解能力增强, 可矿化碳含量下降了47.90%~85.29%;可溶态生物炭被针铁矿吸附后, 可矿化碳含量下降了72.68%~78.76%.由表 2可知, 最不稳定的可溶态生物炭只占整体生物炭的1%, 其中可降解的只有9%, 而高岭石或者针铁矿的存在又使可溶态生物炭可矿化碳含量下降高达85%, 由此进一步证明了生物炭在土壤中可发挥长期固碳作用(Kuzyakov et al., 2014).由此可见, 矿物质吸附有机碳是自然条件下很重要的固碳方式(Meier et al., 1999; Watanabe et al., 2005), 相关研究指出, 氨基酸(Gonod et al., 2006)、酶(Lozzi et al., 2001)、小分子的有机酸(van Hees et al., 2003)及沉积物中的有机质(Kalbitz et al., 2005)都能通过该方式得到稳定.
由矿化速率拟合得到的数据进行计算得到半衰期, 结果显示, 可溶态生物炭被吸附后, 半衰期增大; 且吸附量最大的背景溶液体系相应的半衰期最长, 降解速率最慢.这说明矿物质吸附可溶态生物炭的能力与微生物降解被其吸附的可溶态生物炭的能力有关, 吸附结合力越强, 则抵抗微生物降解能力越强.
对于高岭土, 当3种吸附机理都存在时, 半衰期最长.配体交换使得高岭土体系的可矿化碳含量增高(比较NaCl体系与NaCl+NaH2PO4体系).Ca2+架桥(可矿化碳含量4.71%)发挥的固碳作用不如配位体交换(可矿化碳含量1.82%)和范德华力(可矿化碳含量1.33%), Ca2+在高岭土处理中起抑制效果.但从半衰期的角度分析, Ca2+架桥在高岭土体系中使得可溶态生物炭稳定性得到了增加(比较CaCl2体系与NaCl体系的矿化速率), 这是由于Ca2+架桥的吸附力最大, 其与可溶态生物炭的结合最稳定, 因此, 半衰期最长, 这与相关研究的结论相一致(Mikutta et al., 2007).对于针铁矿, 3种背景溶液对于可溶态生物炭的影响效果与高岭土存在差异, Ca2+存在的背景溶液中, 可矿化碳含量最低且半衰期最短, 矿化速率最大.这表明Ca2+在针铁矿吸附可溶态生物炭的体系中可能还存在其他反应(Ali et al., 1996), 这与3.2节中得到的结论相一致.Ca2+的存在对于针铁矿吸附的可溶态生物炭的抗微生物降解存在负作用.
4 结论(Conclusions)1) 高岭土可通过Ca2+架桥、配体交换和范德华力作用3种机理吸附可溶态生物炭, 在可溶态生物炭浓度较低时以Ca2+架桥为主, 约占吸附总量的65%;高浓度时以范德华力吸附为主, 约占吸附总量的76%.针铁矿的吸附量先升高后下降, 以范德华力为主, Ca2+的存在会抑制针铁矿吸附.对于不同矿物质, 吸附机理存在差异且各种吸附机理所占的比例不相同.
2) 可溶态生物炭被矿物质吸附后可以降低其微生物降解性.通过模型拟合得出, 可溶态生物炭与矿物质结合后, 可矿化碳的总量显著降低, 从9.04%最低可下降至1.33%, 下降了85.3%, 半衰期增大, 矿化速率也有不同程度的降低.矿物质可抑制可溶态生物炭的降解, 促进生物炭稳定性, 提高生物炭的长期固碳效应.
Aiken G R, Hsu-Kim H, Ryan J N. 2011. Influence of dissolved organic matter on the environmental fate of metals nanoparticles and colloids[J]. Environmental Science & Technology, 45(8): 3196–3201.
|
Ali M A, Dzombak D A. 1996. Effects of simple organic acids on sorption of Cu2+ and Ca2+ on goethite[J]. Geochimica et Cosmochimica Acta, 60(2): 291–304.
DOI:10.1016/0016-7037(95)00385-1
|
Amy G L, Sierka R A, Bedessem J, et al. 1992. Molecular size distributions of dissolved organic matter[J]. Journal American Water Works Association, 84(6): 67–75.
|
Arnarson T S, Keil R G. 2000. Mechanisms of pore water organic matter adsorption to montmorillonite[J]. Marine Chemistry, 71(3/4): 309–320.
|
Au K K, Penisson A C, Yang S L, et al. 1999. Natural organic matter at oxide/water interfaces: Complexation and conformation[J]. Geochimica et Cosmochimica Acta, 63(19/20): 2903–2917.
|
Awad Y M, Evgenia B A, Ok Y S, et al. 2012. Effects of polyacrylamide biopolymer and biochar on decomposition of soil organic matter and plant residues as determined by C-14 and enzyme activities[J]. European Journal of Soil Biology, 48: 1–10.
|
Bell M J, Worrall F. 2011. Charcoal addition to soils in NE England: A carbon sink with environmental co-benefits?[J]. Science of the Total Environment, 409: 1704–1714.
DOI:10.1016/j.scitotenv.2011.01.031
|
Catalano J G, Park C, Fenter P, et al. 2008. Simultaneous inner-and outer-sphere arsenate adsorption on corundum and hematite[J]. Geochimica et Cosmochimica Acta, 72: 1986–2004.
DOI:10.1016/j.gca.2008.02.013
|
Cornelissen G, Gustafsson O, Bucheli T D, et al. 2005. Extensive sorption of organic compounds to black carbon coal and kerogen in sediments and soils: Mechanisms and consequences for distribution bioaccumulation and biodegradation[J]. Environmental Science & Technology, 39(18): 6881–6895.
|
Feng X J, Simpson A J, Simpson M J. 2005. Chemical and mineralogical controls on humic acid sorption to clay mineral surfaces[J]. Organic Geochemistry, 36(11): 1553–1566.
DOI:10.1016/j.orggeochem.2005.06.008
|
Ghose S K, Waychunas G A, Trainor T P, et al. 2010. Hydrated goethite (alpha-FeOOH) (100) interface structure: Ordered water and surface functional groups[J]. Geochimica et Cosmochimica Acta, 74(7): 1943–1953.
DOI:10.1016/j.gca.2009.12.015
|
Glaser B, Haumaier L, Guggenberger G, et al. 1998. Black carbon in soils: the use of benzenecarboxylic acids as specific markers[J]. Organic Geochemistry, 29(4): 811–819.
DOI:10.1016/S0146-6380(98)00194-6
|
Goldberg E D. 1985. Black Carbon in the Environment Properties and Distribution[M]. J.Wiley: : 197–198.
|
Gonod L V, Jones D L, Chenu C. 2006. Sorption regulates the fate of the amino acids lysine and leucine in soil aggregates[J]. European Journal of Soil Science, 57(3): 320–329.
DOI:10.1111/ejs.2006.57.issue-3
|
Jones D L, Murphy D V, Khalid M, et al. 2011. Short-term biochar-induced increase in soil CO2 release is both biotically and abiotically mediated[J]. Soil Biology & Biochemistry, 43(8): 1723–1731.
|
Kaiser K, Guggenberger G. 2000. The role of DOM sorption to mineral surfaces in the preservation of organic matter in soils[J]. Organic Geochemistry, 31(7/8): 711–725.
|
Kaiser K, Guggenberger G. 2003. Mineral surfaces and soil organic matter[J]. European Journal of Soil Science, 54(2): 219–236.
DOI:10.1046/j.1365-2389.2003.00544.x
|
Kalbitz K, Schwesig D, Rethemeyer J, et al. 2005. Stabilization of dissolved organic matter by sorption to the mineral soil[J]. Soil Biology & Biochemistry, 37(7): 1319–1331.
|
Kuzyakov Y, Bogomolova I, Glaser B. 2014. Biochar stability in soil: Decomposition during eight years and transformation as assessed by compound-specific C-14 analysis[J]. Soil Biology & Biochemistry, 70: 229–236.
|
Lehmann J, Gaunt J, Rondom M. 2010. Bio-char sequestration in terrestrial ecosystems-a review[J]. Mitig Adapt Start for Glob Change, 11: 403–427.
|
Li F, Cao X, Zhao L, et al. 2014. Effects of mineral additives on biochar formation: Carbon retention stability and properties[J]. Environmental Science & Technology, 48(19): 11211–11217.
|
李力, 刘娅, 陆宇超, 等. 2011. 生物炭的环境效应以及其应用进展[J]. 环境化学, 2011, 30(8): 1411–1421.
|
Liang B, Lehmann J, Solomon D, et al. 2006. Black carbon increases cation exchange capacity in soils[J]. Soil Science Society of America Journal, 70(5): 1719–1730.
DOI:10.2136/sssaj2005.0383
|
Lozzi I, Calamai L, Fusi P, et al. 2001. Interaction of horseradish peroxidase with montmorillonite homoionic to Na+ and Ca2+: effects on enzymatic activity and microbial degradation[J]. Soil Biology & Biochemistry, 33(7/8): 1021–1028.
|
Meier M, Namjesnik-Dejanovic K, Maurice P A, et al. 1999. Fractionation of aquatic natural organic matter upon sorption to goethite and kaolinite[J]. Chemical Geology, 157(3/4): 275–284.
|
Mikutta R, Kleber M, Torn M S, et al. 2006. Stabilization of soil organic matter: Association with minerals or chemical recalcitrance?[J]. Biogeochemistry, 77(1): 25–56.
DOI:10.1007/s10533-005-0712-6
|
Mikutta R, Mikutta C, Kalbitz K, et al. 2007. Biodegradation of forest floor organic matter bound to minerals via different binding mechanisms[J]. Geochimica et Cosmochimica Acta, 71(10): 2569–2590.
DOI:10.1016/j.gca.2007.03.002
|
Namjesnik-Dejanovic K, Maurice P A. 2001. Conformations and aggregate structures of sorbed natural organic matter on muscovite and hematite[J]. Geochimica et Cosmochimica Acta, 65(7): 1047–1057.
DOI:10.1016/S0016-7037(00)00542-1
|
Nguyen B T, Lehmann J, Kinyangi J, et al. 2008. Long-term black carbon dynamics in cultivated soil[J]. Biogeochemistry, 89: 295–308.
DOI:10.1007/s10533-008-9220-9
|
Schmidt M W I, Torn M S, Abiven S, et al. 2011. Persistence of soil organic matter as an ecosystem property[J]. Nature, 478(7367): 49–56.
DOI:10.1038/nature10386
|
Sollins P, Kramer M G, Swanston C, et al. 2009. Sequential density fractionation across soils of contrasting mineralogy: evidence for both microbial-and mineral-controlled soil organic matter stabilization[J]. Biogeochemistry, 96: 209–231.
DOI:10.1007/s10533-009-9359-z
|
Spark K M, Wells J D, Johnson B B. 1997. Sorption of heavy metals by mineral-humic acid substrates[J]. Australian Journal of Soil Research, 35(1): 113–122.
DOI:10.1071/S96010
|
Strauss R, Brummer G W, Barrow N J. 1997. Effects of crystallinity of goethite 2 Rates of sorption and desorption of phosphate[J]. European Journal of Soil Science, 48(1): 101–114.
DOI:10.1111/ejs.1997.48.issue-1
|
van Hees P A W, Vinogradoff S I, Edwards A C, et al. 2003. Low molecular weight organic acid adsorption in forest soils: effects on soil solution concentrations and biodegradation rates[J]. Soil Biology & Biochemistry, 35(8): 1015–1026.
|
Watanabe N, Schwartz E, Scow K M, et al. 2005. Relating desorption and biodegradation of phenanthrene to SOM structure characterized by quantitative pyrolysis GC-MS[J]. Environmental Science & Technology, 39(16): 6170–6181.
|
Weng L P, Koopal L K, Hiemstra T, et al. 2005. Interactions of calcium and fulvic acid at the goethite-water interface[J]. Geochimica et Cosmochimica Acta, 69(2): 325–339.
DOI:10.1016/j.gca.2004.07.002
|
Yang F, Zhao L, Gao B, et al. 2016. The interfacial behavior between biochar and soil minerals and its effect on biochar stability[J]. Environmental Science & Technology, 50: 2264–2271.
|
Zhao L, Cao X, Mašek O, et al. 2013. Heterogeneity of biochar properties as a function of feedstock sources and production temperatures[J]. Journal of Hazardous Materials, 256: 1–9.
|
Zimmerman A R. 2010. Abiotic and microbial oxidation of laboratory-produced black carbon (Biochar)[J]. Environmental Science & Technology, 44(4): 1295–1301.
|
左继超, 胡红青, 刘永红, 等. 2017. 磷和柠檬酸共存对高岭石和针铁矿吸附铅的影响[J]. 土壤学报, 2017: 265–272.
DOI:10.11766/trxb201606160113 |