 2017, Vol. 37
2017, Vol. 37



2. 广东省环境污染控制与修复技术重点实验室, 广州 510275;
3. 广西环境污染控制理论与技术重点实验室, 桂林理工大学环境科学与工程学院, 桂林 541004
2. Guangdong Provincial Key Laboratory of Environmental Pollution Control and Remediation Technology, Guangzhou 510275;
3. Guangxi Key Laboratory of Environmental Pollution Control Theory and Technology, College of Environmental Science and Engineering, Guilin University of Technology, Guilin 541004
近年来, 利用纳米零价铁(nano-scale zero-valent iron, nZVI)对土壤进行原位修复是环境修复领域的研究热点(周启星等, 2001).nZVI是指粒径在1~100 nm范围内的零价铁颗粒.与普通零价铁相比, nZVI比表面积更大, 有利于重金属的吸附; 另一方面, 由于表面效应, nZVI表面原子数迅速增加, 还原性增强; 同时, 当零价铁颗粒尺寸处于纳米量级时, nZVI特殊的理化性质也使其性能明显优于普通零价铁(高圆圆等, 2013).所以nZVI在土壤原位修复方面具有极大的应用潜力.与此同时, 土壤是一种高密度介质, 孔隙较小, 对nZVI的吸附力较强; 且土壤中成分复杂, 影响因子极多, 如土壤pH、腐殖酸、共存离子等都对nZVI的活性有着较大影响(Zhao et al., 2016).因此, nZVI在土壤修复应用中还存在很多问题, 主要包括: ① nZVI自身磁性极强, 同时nZVI粒径小, 比表面积大, 颗粒之间极易团聚在一起, 促使粒径变大, 导致比表面积大幅降低, 反应效率降速; ② nZVI极易被氧化, 从而产生一层致密的氧化物或钝化层覆盖在表面, 活性降低; 甚至nZVI的快速氧化会发生自燃现象, 这些情况极大的限制了nZVI在地下水和土壤修复中的应用; ③ nZVI具有生物毒性, 高浓度的nZVI对生物具有杀灭作用(舒中亚等, 2011); 同时在应用中, nZVI难回收、易流失、容易造成二次污染; 以上这些问题很大程度上限制了nZVI在土壤修复中的实际应用.
针对这些问题, 研究人员探索尝试了多种方法强化nZVI在环境中移动性与稳定性.为了抑制纳米颗粒的团聚效应和保持其在环境中的反应活性与迁移能力, 越来越多的nZVI强化技术应运而生.而其中对nZVI进行表面修饰的方法成为目前研究最多的方法之一(冯倩微等, 2014).nZVI的表面改性手段主要分为两大类: ①加入表面活性剂; ②加入聚合物高分子电解质.这两种方式都是通过静电斥力或空间位阻作用减少纳米颗粒团聚效应, 从而达到增强颗粒稳定活性之目的(庞龙等, 2011).Lin等(2010)用聚乙烯丙烯酸(PAA)和羧甲基纤维素(CMC)对nZVI进行表面修饰, 结果表明, 修饰后的粒子在多孔介质中的传输速率明显加快, 猜想其原因为经PAA与CMC修饰后的粒子表面含有大量羧基, 很大程度上增加了纳米颗粒间的静电斥力以及空间位阻作用, 很好的抑制了nZVI间团聚, 保持其稳定性, 提高了其降解性能.因此, 本研究选用CMC-nZVI作为供试稳定型纳米零价铁材料.
目前, 室内和室外试验研究已经表明稳定化nZVI能够有效降解卤代有机物和固化有毒金属、类金属和放射性核素(Zhao et al., 2016).然而, 由于土壤理化性质的复杂性, 受污染金属的多样性, nZVI对不同金属离子的固定机制也不尽相同; 现阶段采用nZVI原位修复重金属污染土壤的研究以修复场地Cr(Ⅵ)污染土壤的报道居多(Jiemvarangkul et al., 2011; Wang et al., 2014), 对Cr(Ⅵ)污染土壤固定机制最为了解, 但其中的固定机制仍存在分歧, 如Gao等(2006)认为Cr(Ⅵ)会被nZVI还原为Cr(Ⅲ), 然后以氢氧化物沉淀的形式赋存于土壤表面.而Chrysochoou等(2012)则暗示吸附和共沉淀可能是nZVI去除Cr(Ⅵ)的原因.因此, 在大规模实地应用nZVI修复技术之前掌握不同土壤中有毒金属的修复机制是很有必要的.在矿山开采过程中, 矸石山、尾矿场、露天采矿场、排土场、塌陷区等因重金属污染而失去经济利用价值的土地称为矿山废弃地(黄铭洪, 等. 2003).矿山废弃地具有以下特点: ①物理结构不良, 持水保肥能力差; ②极端贫瘠, N、P、K及有机质含量极低; ③重金属含量过高; ④常为高硫土壤, 间接导致极酸的土壤pH环境.因此, 矿山废弃地不仅破坏和侵占了大量土地资源, 同时也会带来严重的生态环境问题(Dudka and Adriano, 1997; Sasaki et al., 2002).大宝山位于广东省韶关市, 是一座以铁铜为主的大型多金属矿山.大宝山污染土壤, 有机质贫瘠、硫含量高和铜(Cu)污染严重(周永章等, 2008).铜的亲硫性极强, 土壤中含铁矿物和硫酸盐的变化, 强烈影响着铜在土壤中存在形态(李义纯等, 2009).本文研究CMC-nZVI对大宝山矿山高硫富铜土壤中Cu-Fe-S的形态转化与环境行为, 探讨CMC-nZVI对高硫土壤中Cu的固定修复机制, 为改性nZVI在矿山污染土壤修复工作中的应用提供科学依据.
2 材料与方法(Materials and methods) 2.1 供试土壤本研究所用的土壤样品, 釆自大宝山矿区排土场, 地理位置22.55°N, 113.72°E, 海拔720 m; 占地约2 hm2, 位于大宝山凡洞矿山附近.土壤采集使用梅花形采样法, 研究所用土壤为0~20 cm表层土壤样品, 实验土壤的基本理化性质如表 1所示.据表 1可知, 供试土壤酸性强, 有机质含量低, 且土壤中Cu的含量已超过了土壤环境质量标准(GB15618—1995)中国家Ⅲ类土壤标准限值(Cu≤400 mg·kg-1).
| 表 1 土壤基本理化性质(平均值±标准差, n=3) Table 1 Soil basic properties (mean ± SD, n=3) |
本实验中稳定型nZVI的制备采用硼氢化钠还原法(Wang et al., 2010).首先, 称取0.278 g FeSO4·7H2O, 溶于8 mL水和2 mL无水乙醇中.然后, 制备0.1% CMC溶液, 称取0.1 g羧甲基纤维素钠(CMC)溶于80 mL水和20 mL无水乙醇中.将FeSO4溶液与CMC溶液混合, 转移入250 mL带支孔的锥形瓶中, 通N2, 磁力搅拌15 min, 得到Fe2+-CMC混合溶液.而后在磁力搅拌和持续通N2的条件下, 通过分液漏斗缓慢滴加一定浓度的NaBH4溶液, 即可得到黑色的nZVI悬浊液.所得的稳定型nZVI通过外加磁力将其分离收集, 真空冷冻干燥.制备过程中所用水为高纯水, 并通入15 min N2去除O2.
2.3 实验设计因低浓度乙醇可促进厌氧微生物的活性(Lovley and Phillips, 1986), 故通N2 15 min去氧配制1.456‰乙醇溶液.然后, 称取一定量的CMC-nZVI粉末, 配制浓度为1 g·L-1的CMC-nZVI乙醇悬浊液.通N2, 而后盖紧试剂瓶瓶塞, 超声分散2 min, 备用.
实验采用一系列100 mL聚乙烯离心管, 分别加入15 g大宝山污染土壤.向离心管中快速转移75 mL 1 g·L-1 CMC-nZVI乙醇悬浊液(土:溶液=1 g: 5 mL), 继而用N2顶空吹扫后拧紧盖子密封, 摇匀, 而后置于摇床中200 r·min-1, 振荡12 h.取出后, 垂直放置于室内.分别于第1、3、5、10、15、20 d取样、分析和测试.空白对照组则用1.456‰乙醇溶液代替CMC-nZVI乙醇溶液, 其他实验方法与处理组相同.所有处理均重复3次.
2.4 分析方法 2.4.1 水相分析方法为使水土分离, 便于取样, 取样前将样品离心管以3000 r·min-1离心6 min, 上清液用0.45 μm的水系滤膜过滤, 采用HNO3消解后, 使用电感耦合等离子发射光谱仪ICP-OES(5300DV, Perkin Elmer, USA)测定消解液中Cu的浓度.水相中的SO42-使用离子色谱仪(Ion chromatography-732, 万通, 瑞士)进行检测(最低检出限为2.0 mg·L-1); 水相中亚铁离子(Fe2+)浓度采用邻菲罗啉分光光度法进行检测.仪器分析测试过程严格遵从质量控制原则, 相关分析测试指标所用标准曲线要求R2达到0.999以上方可进行样品检测, 同时每测试20个样品必须校准1个标准物质样品, 在合理偏差之内则继续测试样品, 下同.
2.4.2 固相分析方法① TCLP提取实验:为了评估CMC-nZVI对大宝山Cu污染土壤的修复效果, 采用毒性淋溶提取试验(Toxicity characteristic leaching procedure, TCLP)考察处理前后的土壤中Cu的滤出性.根据有毒物质溶出国际标准方法(US EPA Method 1311), 本实验所用土壤应采用提取液#1(5.7 mL冰醋酸加入到500 mL试剂水, 再加入64.3 mL 1 mol·L-1的NaOH, 稀释到1 L, 此溶液pH为(4.93±0.05)进行提取.参照美国环保署EPA1311方法, 将离心后的土样和提取液按1 g:20 mL比例混合, 置于旋转振荡器上以30 r·min-1室温下振荡18 h, 离心分离.上清液用0.45 μm的微孔滤膜过滤, 滤液消解后, 样品消解液中Fe、Cu浓度采用电感耦合等离子发射光谱仪ICP-OES(5300DV, Perkin Elmer, USA)进行测定(最低检出限都为0.002 mg·L-1).
② 重金属形态测定:土壤重金属总量消解方法采用土壤环境质量标准(GB15618—1995)的湿法消解.取过100目尼龙筛后的干燥土壤约0.5~1.0 g, 加入盐酸、硝酸与高氯酸(3:1:1)于300 ℃消解, 消解、过滤后使用5%硝酸定容待测.同时采用传统Tessier五态连续提取的改良方法(Guo et al., 2009), 将重金属形态分为S1弱酸提取态, S2铁锰氧化物结合态, S3有机结合态, S4硫化物结合态, S5残渣态进行连续提取.对第1、3、5、10、15、20 d土样中重金属形态进行五态分析.土壤中Fe和Cu的含量用ICP-OES(5300DV, Perkin Elmer, USA)进行测定.
③ 土壤中总S和酸可挥发性硫化物的测定方法:土壤中总S的测试采用燃烧碘量法(GB7875-87);土壤样品的酸可挥发性硫化物(acid-volatile sulfides, AVS)提取采用冷扩散吸收方法(Hsieh et al., 2002), 硫化物测定方法为水质硫化物测定亚甲基蓝分光光度法(GB/T16489—1996).
④ XRD分析: X射线衍射(X-ray diffraction, XRD)是利用晶体形成的X射线衍射, 对物质进行内部原子在空间分布状况的结构分析方法.为研究施加CMC-nZVI前后土壤环境中含Cu矿物变化情况, 故采用XRD分析技术对第1 d和第20 d的1 g·L-1 CMC-nZVI处理的土壤样品进行物相检测.采用的仪器为荷兰帕纳科公司生产的Empyrean(锐影)X射线衍射仪.测试条件为:功率3 kW; 最小扫描步长0.02°; 测角范围(2θ)为10°~80°.
2.4.3 数据处理方法用Jade 6.0软件, 进行XRD物相检索和鉴定.用Microsoft Excel 2007和Origin 8.0处理数据及图表, 用SPSS 12.0统计软件进行试验数据的统计分析.
3 结果(Results) 3.1 CMC-nZVI对高硫土壤中Cu的固定效果由图 1可知, 未添加CMC-nZVI处理的土壤, Cu的浸出浓度超过EPA规定的15 mg·kg-1, 属于危险废物(Sun et al., 2006).在0~20 d内, 1 g·L-1 CMC-nZVI处理Cu的浸出浓度均低于15 mg·kg-1, 达到安全标准.
 |
| 图 1 不同修复时间下两组土壤样品中Cu的TCLP浸出浓度 Fig. 1 TCLP leachability of Cu under different treatments at various treatment time |
由图 2可知, CMC-nZVI处理第1 d后, 土壤中弱酸提取态(Cu-S1)含量较对照组大幅降低了63%(p < 0.05);与之不同, 土壤中硫化物结合态(Cu-S4)含量则显著高于对照组2倍左右(p < 0.05).
 |
| 图 2 不同修复时间下土壤中铜的结合形态的变化(a.对照组, b.CMC-nZVI处理组)(S1弱酸提取态, S2铁锰氧化物结合态, S3有机结合态, S4硫化物结合态, S5残渣态, 下同) Fig. 2 Changes of Cu speciation in soils at various treatment times (a. control, b. CMC-nZVI group) |
随着培养时间的延长, 对照组土壤中铁锰氧化物结合态Cu-S2和有机结合态(Cu-S3)的含量逐渐降低; 从第10 d开始, 对照组土壤中Cu-S1和Cu-S4的含量逐渐升高(图 2).然而, CMC-nZVI处理组土壤中Cu的5种赋存形态的含量基本趋于稳定(p>0.05).
3.3 土壤中总Fe的形态变化过程由图 3可知, 与对照组相比, 添加CMC-nZVI处理后土壤淋溶液中亚铁和总铁浓度都明显更高.添加CMC-nZVI处理后淋溶液中Fe2+浓度表现为第0~10 d波动剧烈, 10 d后基本保持稳定.添加CMC-nZVI后淋溶液中总Fe浓度整体升高, 前5 d淋溶液中总Fe浓度大幅增加, 10 d后总Fe浓度趋于稳定.
 |
| 图 3 不同处理水相中Fe2+(a)及总Fe(b)分布变化情况 Fig. 3 Distribution of (a) Fe2+ and (b) Fe-Total in aqueous phase under different treatments |
由图 4可知, 与对照组不同, 添加CMC-nZVI后土壤固相中Fe-S1、Fe-S3含量随时间增加, Fe-S2、Fe-S4含量基本保持稳定.一方面, 矿物溶解及CMC-nZVI氧化, 导致水相Fe(Ⅲ)浓度增加, 而Fe(Ⅲ)水解生成氢氧化铁Fe(OH)3和水合氧化铁FeOOH, 通过悬浮、结合、絮凝, 沉降到固相中, Fe-S1含量增加.而伴随CMC-nZVI的氧化, 其颗粒表面氧化膜厚度不断增加, 且Fe3O4, γ-Fe2O3等结晶性铁氧化物含量增加(Dumitrache et al., 2005); 同时, nZVI氧化和铁矿物的还原溶解向水相中释放大量Fe(Ⅱ), Fe(Ⅱ)能与过量的Fe(Ⅲ)形成混合价态的FeⅡFe2ⅢO4(Muehe et al., 2013), 这些过程直接导致Fe-S2含量增加补充了铁还原菌对铁氧化物的消耗, 所以Fe-S2含量基本保持稳定, 与对照组变化不同; 同时CMC-nZVI不仅可以作为电子供体, 并且其表面的CMC能够为微生物提供碳源(颜霞等, 2008), 补充土壤中SOM的消耗, 所以Fe-S3含量随时间略微增加, 而对照组由于微生物消耗大量碳源, 体系有机质含量降低, Fe-S3含量随时间延长而降低.CMC-nZVI处理组第1 d时, Fe-S4含量远高于对照组, 且观察期内基本保持稳定.同时Fe-S5相对于对照组也较高, 这可能是Fe-S4向Fe-S5转化所造成, 因为Fe-S4可能进一步矿化为黄铁矿FeS2(吴锡等, 2012).
 |
| 图 4 不同修复时间下土壤中铁的结合形态的变化(a.对照组, b. CMC-nZVI处理组) Fig. 4 Changes of Fe speciation in soils at various treatment times(a. control, b. CMC-nZVI group) |
Fe-S4、Fe-S5的变化与体系中硫元素的变化密切相关, 纳米零价铁及微生物的联合作用导致体系中产生大量S2-将体系中游离的Fe(Ⅱ)以FeS或FeS2等形式沉淀下来(Burton, 2010), 导致Fe-S4、Fe-S5含量增加, 具体过程将在下一节具体阐述.
3.4 土壤中SO42-的形态变化过程由图 5可知, 添加CMC-nZVI后, 水相中SO42-浓度明显低于对照组.0~10 d上清液中SO42-浓度逐渐升高, 10 d后, 上清液中SO42-浓度再次逐渐降低.相应的, 添加CMC-nZVI初期, 土壤中酸可挥发硫化物AVS含量增加.CMC-nZVI加入土壤后, 体系转为还原环境, SO42-易得到电子, 被还原为S2-, 而S2-易与Fe、Cu形成硫化物沉淀, 从而导致体系AVS含量增加(郭鹏然等, 2010).CMC-nZVI处理土壤中, 1~5 d AVS含量不断降低, 主要是因为纳米零价铁不断被氧化, 引起硫化物向硫酸盐转化(Steven et al., 2005).此时对照组未检测出AVS的存在.从第5 d开始, CMC-nZVI处理中AVS的含量再次增加.而对照组从第10 d开始AVS含量增加.这是因为添加低浓度乙醇, 作为电子供体, 促进厌氧微生物(Lovley and Phillips, 1986), 如硫酸盐还原菌活性增强, 加速硫酸根还原过程.
 |
| 图 5 不同处理水相中SO42-(a)及固相中AVS(b)的变化趋势 Fig. 5 Content changes of (a) SO42- in aqueous phase and (b) AVS in solid phase under different treatments |
而CMC-nZVI处理组, 硫酸盐还原菌从第5 d开始活性增强, 早于对照组.有研究表明, 纳米零价铁可以促进硫酸盐还原菌的生长(Barnes et al., 2010).5 d后, 1 g·L-1 CMC-nZVI处理土壤中pH及Eh基本保持稳定, 为硫酸盐还原菌的生长创造适宜的环境(颜晓元等, 1996).另一方面, nZVI还原所产生的氢气, 大多数SRB以H2为电子供体, 促进了SRB的活性; 同时, Fe(Ⅱ)在酸性高硫环境中Fe-S的还原过程起着触发器的作用(Burton et al., 2008); 水相中Fe2+增加, 一方面促进细菌细胞的生长, 另一方面与反应生成的硫化氢形成沉淀, 消除硫化氢对SRB的抑制作用, 增强了SRB的活性(Fan et al., 2013).
4 讨论(Discussion)由图 1可知, 第1 d时, 1 g·L-1 CMC-nZVI处理淋滤液中Cu的浓度为8.34 mg·kg-1, 与未添加CMC-nZVI的处理相比下降了63%.第1~5 d土壤淋滤液中Cu的浓度上升, 而5~20 d后降低, 20 d Cu的浸出浓度为6.34 mg·kg-1, 稍低于第一天时的浓度.这可能是因为0~5 d CMC-nZVI逐渐被氧化, 对Cu的固定效果变差; 而后期厌氧微生物活性逐渐增强, 强化了对Cu的固定效果(Fan et al., 2013).
由图 4可知, 对照组中Fe-S2(铁锰结合态)含量降低, 20 d时与第1 d相比约降低了50%;淋溶液中总Fe浓度升高, 且10 d后水相中产生Fe2+(图 3), 这可能是因为在微生物作用下铁锰氧化物发生了还原溶解(颜晓元等, 1996).另有研究表明, 在还原土壤中铁氧化物及硫化物的变化强烈影响重金属的活性变化(Muehe et al., 2013).一方面, 土壤淹水后, 随着体系还原程度增强, 铁氧化物发生还原溶解, 导致吸附在其表面的重金属释放; 另一方面, 铁氧化物发生还原溶解的同时伴随新的次级矿物产生, 对重金属的活性也产生重要影响(Muehe et al., 2013).在微生物作用下, 体系中硫酸盐还原形成硫离子, 因CuS的溶解度极低(约为4.0×10-38)可直接形成Cu的硫化物沉淀, 或者通过形成Fe的硫化物以吸附或共沉淀的方式固定Cu(吴锡等, 2012).在此过程中, 微生物活动必不可少, 有机质作为电子供体及微生物活动的碳源被大量消耗(Lovley and Phillips, 1986).所以本研究对照组Cu的形态变化表现为S2、S3含量降低, S4含量增加(图 2).而CMC-nZVI的添加导致土壤中Cu的结合形态具有明显不同的变化过程.与对照组相比, 添加CMC-nZVI处理后致使土壤固相中S2、S3、S4较为稳定的形态含量明显增加(p < 0.05), 且为期20 d的实验过程中这些形态的含量基本保持稳定(p>0.05).
此外, 通过检测土壤pH变化情况可知, 实验第1天时对照组土壤pH为3.79, 而添加1 g·L-1 CMC-nZVI的处理组的土壤pH为5.21, 土壤的极酸性有明显提高和改善.这可能是因为纳米零价铁能够与土壤体系中的水反应生成氢气和氢氧根离子造成的(Yan et al., 2010), 如反应式(1)和(2)所示.

|
(1) |
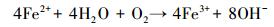
|
(2) |
但随着处理时间的延长, 添加CMC-nZVI处理的pH均呈现出下降的趋势, 这主要是因为纳米零价铁在体系中反应生成Fe(Ⅱ)及Fe(Ⅲ), 而Fe(Ⅱ)及Fe(Ⅲ)的水解会直接导致体系pH值降低, 详见反应式(3)和(4) (Yan et al., 2010).由监测数据可知(详细数据略), 在处理的0~10 d内土壤pH下降速度较快, 处理10 d后, 处理组的土壤pH基本保持稳定.其中1 g·L-1 CMC-nZVI处理的土壤pH为4.06.
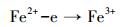
|
(3) |
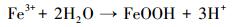
|
(4) |
通过检测土壤Eh变化情况可知, 添加CMC-nZVI进入土壤导致, 土壤Eh值迅速降低.1 g·L-1 CMC-nZVI加入土壤后第1 d土壤中Eh值为-529 mV.这主要是由于纳米零价铁的强还原性, 强烈影响着体系的Eh值; 随着土壤中纳米零价铁的逐渐氧化和消耗, 体系Eh值不断升高.从第5 d开始, 1 g·L-1 CMC-nZVI处理组的土壤Eh值开始高于0 mV, 而后保持120 mV左右趋于稳定状态.一方面, 由于CMC-nZVI有较大的比表面积, 加入土壤后, CMC-nZVI将游离的Cu(Ⅱ)吸附至表面; 另一方面, CMC-nZVI具有强还原性, Cu(Ⅱ)部分被还原为Cu(I), 产生Cu2O (Shi et al., 2013).从我们的XRD分析结果可知(图 6), 添加CMC-nZVI处理后第1 d土壤的XRD谱图中出现Fe(0)及Cu2O的特征峰, 与前人研究结果一致(Shi et al., 2013).
 |
| 图 6 CMC-nZVI处理组不同反应时间的土壤的XRD谱图 Fig. 6 XRD spectrum of CMC-nZVI treated soil samples at different reaction times |
研究体系中AVS和Fe、Cu硫化物结合态的增加, 说明CMC-nZVI加入土壤后, 体系转为还原环境(处理土壤第1 d Eh= -529 mV), SO42-易得到电子, 被还原为S2-, 而S2-易与Fe、Cu形成硫化物沉淀, 使得体系中游离的Cu(Ⅱ), 一方面与新生成的Fe的硫化物(FeS及FeS2)以吸附或共沉淀的方式固定重金属, 或者直接产生Cu的硫化物沉淀(吴锡等, 2012), 将其固定.我们的XRD谱图中鉴定出Cu3KS2等硫化物特征峰的存在为此提供了直接证据(图 6).随着CMC-nZVI被氧化, 其还原能力降低, 吸附变为主要的固定机理(Crane et al., 2011).随着时间的延长, 铁矿物的存在状态也发生极大的变化.由于CMC-nZVI的氧化, 体系pH降低, 同时微生物的活性增强, 导致大量铁矿物被还原溶解, 同样引起Cu(Ⅱ)的活化释放(Muehe et al., 2013).然而伴随着铁矿物的溶解, 新生成的铁氧化物的无定形或微晶形结构能大量吸附溶液中的重金属离子(Tack et al., 2006).体系中新生成的无定形或微晶型次级铁矿物易通过吸附或共沉淀将游离的Cu(Ⅱ)固定.另外, 微生物也会强烈影响CMC-nZVI的存在状态(Wang and Tseng, 2009).在20 d土壤样品的谱图中, 仍可检测出α-Fe0的特征峰存在.体系中活跃的铁还原菌可以导致体系中铁氧化物的分解, 对于CMC-nZVI而言, 可能也会分解其表面的铁氧化物钝化薄膜, 增强其活性(Wang and Tseng, 2009).
同时, CMC-nZVI可以促进硫酸盐还原菌的生长.在CMC-nZVI的作用下, 硫酸盐还原菌等微生物活性极强, 导致硫酸盐重新被还原为S2-, 更多的Cu(Ⅱ)以硫化物的形式被固定下来.所以在20 d的谱图中依然存在CuS、Na2S等硫化物特征峰.
5 结论(Conclusions)CMC-nZVI对高硫土壤中的Cu固定效果显著, 极大地降低了矿山废弃土壤的环境风险.CMC-nZVI施加入土壤后, 土壤中硫化物结合态的Fe含量显著增加; 土壤中AVS的含量也明显增加; 相应Cu硫化物结合态含量明显增加.结合XRD矿物分析结果, CMC-nZVI对Cu固定可能包括以下几个过程: ①CMC-nZVI直接对重金属的吸附与还原; ②施加CMC-nZVI后, 土壤体系变为强还原体系, 大量存在SO42-被还原为S2-, 通过FeS吸附或直接产生硫化物沉淀, 加强了CMC-nZVI对Cu的固定效果; ③ CMC-nZVI可以与体系中厌氧微生物联合作用, 还原溶解铁锰氧化物, 促进次级铁矿物的产生, 同时加速硫酸根还原, 通过产生硫化物沉淀或者与次级铁矿物结合、共沉淀的方式固定Cu, 从而强化维持体系中重金属的固定效果.同时微生物可以影响CMC-nZVI的表面结构, 增强其活性.所以CMC-nZVI可以与土壤中厌氧微生物产生协同作用, 强化对高硫土壤重金属固定效果.
Barnes R J, van C J, Riba O, et al. 2010. The impact of zero-valent iron nanoparticles on a river water bacterial community[J]. Journal of Hazardous Materials, 184(1/3): 73–80.
|
Burton E D. 2010. Effect of sample pretreatment on the fractionation of Fe, Cr, Ni, Cu, Mn, and Zn in acid sulfate soil materials[J]. Geoderma, 159: 156–164.
DOI:10.1016/j.geoderma.2010.07.007
|
Burton E D, Bush R T, Sullivan L A, et al. 2008. Schwertmannite transformation to goethite via the Fe(Ⅱ) pathway: Reaction rates and implications for iron-sulfide formation[J]. Geochimica et Cosmochimica Acta, 72(18): 4551–4564.
DOI:10.1016/j.gca.2008.06.019
|
Cao J S, Zhang W X. 2006. Stabilization of Chromium ore processing residue (COPR) with nanoscale iron particles[J]. Journal of Hazardous Materials, B132: 213–219.
|
Chrysochoou M, Johnston C P, Dahal G. 2012. A comparative evaluation of hexavalent chromium treatment in contaminated soil by calcium polysulfide and green-tea nanoscale zero-valent iron[J]. Journal of Hazardous Materious, 201-202: 33–42.
DOI:10.1016/j.jhazmat.2011.11.003
|
Crane R A, Dickinson M, Popescu I C, et al. 2011. Magnetite and zero-valent iron nanoparticles for the remediation of uranium contaminated environmental water[J]. Water Research, 45(9): 2931–2942.
DOI:10.1016/j.watres.2011.03.012
|
Dudka S, Adriano D C. 1997. Environmental impacts of metal ore mining and processing: a review[J]. Journal of Environmental Quality, 26(3): 590–602.
|
Dumitrache F, Morjan I, Alexandrescu R, et al. 2005. Iron oxide core shell nanoparticles synthesized by laser pyrolysis followed by superficial oxidation[J]. Applied Surface Science, 247(1/4): 25–31.
|
Fan D M, Roberto P A, Bradley M T, et al. 2013. Reductive sequestration of pertechnetate (99TcO4-) by nano zerovalent iron (nZVI) transformed by abiotic sulfide[J]. Environmental Science and Technology, 47: 5302–5310.
DOI:10.1021/es304829z
|
冯婧微, 徐英侠, 兰希平, 等. 2014. 纳米零价铁的改性及其应用研究进展[J]. 材料导报, 2014, 28(15): 83–86.
|
Guo P R, Mou D H, Wang C, et al. 2009. Research of sequential extraction procedure for heavy metals in sediments from mariculture area[J]. Chinese Journal of Analytical Chemistry, 37(11): 1645–1650.
|
郭鹏然, 仇荣亮, 牟德海, 等. 2010. 珠江口桂山岛沉积物中重金属生物毒性评价和同步萃取金属形态特征[J]. 环境科学学报, 2010, 30(5): 1079–1086.
|
高园园, 周启星. 2013. 纳米零价铁在污染土壤修复中的应用与展望[J]. 农业环境科学学报, 2013, 32(3): 418–425.
|
Hsieh Y P, Chung S W, Tsau Y J, et al. 2002. Analysis of sulfides in the presence of ferric minerals by diffusion methods[J]. Chemical Geology, 182(2): 195–201.
|
黄铭洪, 骆永明. 2003. 矿区土地修复与生态恢复[J]. 土壤学报, 2003, 40(2): 161–169.
DOI:10.11766/trxb200210100201 |
Jiemvarangkul P, Zhang W, Lien H L. 2011. Enhanced transport of polyelectrolyte stabilized nanoscale zero-valent iron (nZVI) in porous media[J]. Chemical Engineering Journal, 170(2): 482–491.
|
Lin Y H, Huihsin T, Mingyen W, et al. 2010. Characteristics of two types of stabilized nano zero-valent iron and transport in porous media[J]. Science of Total Environment, 408(10): 2260–2267.
DOI:10.1016/j.scitotenv.2010.01.039
|
Lovley D R, Phillips E J. 1986. Organic matter mineralization with reduction of ferric iron in anaerobic sediments[J]. Journal of General Microbiology, 51(4): 683–689.
|
李义纯, 葛滢. 2009. 淹水还原条件下土壤铁氧化物对镉活性制约机理的研究进展[J]. 土壤, 2009, 41(2): 160–164.
|
Muehe E M, Adaktylou I J, Obst M, et al. 2013. Organic carbon and reducing conditions lead to cadmium immobilization by secondary Fe mineral formation in a pH-neutral soil[J]. Environmental Science & Technology, 47(23): 3092–3092.
|
庞龙, 周庆祥, 苏现伐. 2011. 纳米零价铁修饰技术研究进展[J]. 化工进展, 2011, 30: 1361–1368.
|
Sasaki K, Haga T, Hirajima T, et al. 2002. Distribution and transition of heavy metals in mine tailing dumps[J]. Materials Transactions, 43(11): 2778–2783.
DOI:10.2320/matertrans.43.2778
|
Shi L N, Zhou Y, Chen Z, et al. 2013. Simultaneous adsorption and degradation of Zn2+ and Cu2+ from wastewaters using nanoscale zero-valent iron impregnated with clays[J]. Environmental Science and Pollution Research, 20(6): 3639–3648.
DOI:10.1007/s11356-012-1272-7
|
Steven W, Charnock J M, John H, et al. 2005. Sulfide species as a sink for mercury in lake sediments[J]. Environmental Science Technology, 39(17): 6644–6648.
DOI:10.1021/es048874z
|
Sun Y, Xie Z, Li J, et al. 2006. Assessment of toxicity of heavy metal contaminated soils by the toxicity characteristic leaching procedure[J]. Environmental Geochemistry & Health, 28(1/2): 73–78.
|
舒中亚, 汪杰, 黄艺. 2011. 零价铁纳米颗粒对硫酸盐还原菌的杀灭作用研究[J]. 环境科学, 2011, 32: 3040–3044.
|
Tack F M G, Ranst E V, Lievens C, et al. 2006. Soil solution Cd, Cu and Zn concentrations as affected by short-time drying or wetting: The role of hydrous oxides of Fe and Mn[J]. Geoderma, 137(1/2): 83–89.
|
Wang Q, Qian H, Yang Y, et al. 2010. Reduction of hexavalent chromium by carboxymethyl cellulose-stabilized zero-valent iron nanoparticles[J]. Journal of Contaminant Hydrology, 114(1): 35–42.
|
Wang S M, Tseng S K. 2009. Dechlorination of trichloroethylene by immobilized autotrophic hydrogen-bacteria and zero-valent iron[J]. Journal of Bioscience and Bioengineering, 107(3): 287–292.
DOI:10.1016/j.jbiosc.2008.11.010
|
Wang Y, Fang Z Q, Kang Y, et al. 2014. Immobilization and phytotoxicity of chromium in contaminated soil remediated by CMC-stabilized nZVI[J]. Journal of Hazardous Materious, 275: 230–237.
DOI:10.1016/j.jhazmat.2014.04.056
|
吴锡, 许丽英, 张雪霞, 等. 2012. 缺氧条件下土壤砷的形态转化与环境行为研究[J]. 环境科学, 2012, 33(1): 273–279.
|
Yan W L, Herzing A A, Kiely C J, et al. 2010. Nanoscale zero-valent iron (nZVI): aspects of the core-shell structure and reactions with inorganic species in water[J]. Journal of Contaminant Hydrology, 118(3/4): 96–104.
|
颜晓元, 蔡祖聪. 1996. 淹水土壤中甲烷产生的影响因素研究进展[J]. 环境科学进展, 1996, 4(2): 24–32.
|
颜霞, 秦魏. 2008. 降解纤维素真菌的分离筛选及其环境适应性初探[J]. 中国农学通报, 2008, 24(5): 44–49.
|
Zhao X, Liu W, Cai Z Q, et al. 2016. An overview of preparation and applications of stabilized zero-valent iron nanoparticles for soil and groundwater remediation[J]. Water Research, 100: 245–266.
DOI:10.1016/j.watres.2016.05.019
|
周永章, 付善明, 张澄博, 等. 2008. 华南地区含硫化物金属矿山生态环境中的重金属元素地球化学迁移模型——重点对粤北大宝山铁铜多金属矿山的观察[J]. 地学前缘, 2008, 15(5): 248–255.
|
周启星, 林海芳. 2001. 污染土壤及地下水修复的PRB技术及展望[J]. 环境污染防治技术与设备, 2001, 2(5): 48–53.
|