 2017, Vol. 37
2017, Vol. 37



化学氧化处理技术对废水中具有毒性且难以生物降解的有机污染物的治理效果显著.其中高级氧化技术(Advanced Oxidation Technologies, AOTs)是最有效的化学氧化技术之一, 在实际废水的处理中发挥了重要作用(马龙等, 2016).在AOTs中, 反应遵循特有的氧化途径, 通常包括形成高活性的自由基, 如· OH或SO4-·.这些自由基的化学活性很高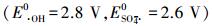
由于这些局限, 科学家和工程师们一直致力于开发和应用各种改进的Fenton技术及新型高级氧化技术.近年发展起来的基于硫酸根自由基(SO4-·)的新型高级氧化技术对有机污染物的去除效果好, 且适应范围广.该技术以单过硫酸盐或者过二硫酸盐为氧化剂, 使用过渡金属Co2+、Ag+、Cu2+、Fe2+及紫外辐射、O3、热等物质为催化剂, 产生高活性的硫酸根自由基SO4-·用于包括除草剂、杀虫剂、环境内分泌干扰物、偶氮染料、酚类物质等多种有机污染物的高效去除(Mielke et al., 2009; 欧阳磊等, 2013;Oh et al., 2015;刘元坤等, 2017).而使用非均相催化剂不仅利于催化剂的分离和重复利用, 且不少固体催化剂的效果可以媲美甚至超过一般的均相催化体系.例如, Pu等(2014)研究了Fe/S杂化的活性炭颗粒高效催化过硫酸盐降解甲基橙和邻苯二甲酸二乙酯;Duan等(2015)对比研究了使用单壁碳纳米管、介孔碳CMK-8及还原型氧化石墨烯活化过硫酸盐高效降解去除酚类和染料污染物的效果, 发现这些非金属催化剂的效果比传统的金属氧化物(Fe3O4、CuO、Co3O4、MnO2)效果更好.当前该高级氧化技术主要围绕新型催化剂的制备和活化机理, 及污染物去除机理的探索而展开.在传统催化剂和催化方式的基础上, 非金属物质及天然有机物作为活化剂, 引起了广泛的关注和研究(Ocampo, 2009; Fang et al., 2013, Lee et al., 2015).这类催化剂不仅成本更低, 环境友好且高效, 来源广泛, 更利于高级氧化技术在原位修复中的应用.
新型的碳材料如活性炭、碳纳米管、碳纤维、富勒烯、氧化石墨烯因其独特的结构和化学特性, 在环境污染物治理领域展示了良好的性能.比如, 庞娅对磁性碳纳米管及其复合材料在水体重金属污染的修复进行了较为系统的研究(庞娅, 2012);使用活性炭作为吸附剂或者催化剂也引起了广泛的关注(李湘等2006;Lee et al., 2015).而近年来对氧化石墨烯的环境应用研究也越来越多, 比如:张琼等(2010)研究了氧化石墨烯复合材料光催化降解有机物的效果;使用改性氧化石墨烯作为吸附剂, 用于环境污染物的去除和痕量污染物的富集分析也被多次报道(肖蓝等, 2013).氧化石墨烯优异的电子传递能力, 丰富的功能基团和表面及边缘的缺陷结构使其在光催化、化学催化氧化中的应用前景广阔(Zhang et al., 2011).而通过对其进行化学修饰, 如非金属元素氮、磷、硫的掺杂, 和金属氧化物形成复合材料更是拓宽了该材料的应用(赵艳丽, 2016).Wang等的研究表明:氮掺杂氧化石墨烯后, 其对BPA的吸附容量是无掺杂的氧化石墨的1.75倍, 其活化过硫酸盐降解BPA的反应速率常数是普通氧化石墨烯的700倍, 降解效率显著提高(Wang et al., 2015).使用单独的纳米Fe3O4或者磁性纳米复合材料作为催化剂在高级氧化技术中的应用研究也得到了较全面的研究.磁性纳米Fe3O4不仅具有大的比表面积, 活性位点多, 且磁性特性使材料作为催化剂的时候, 易于从非均相体系中分离出来, 便于操作和重复利用.而本研究中拟制备磁性氮掺石墨烯作为活化剂, 旨在将氮掺石墨烯的独特性能和磁性纳米Fe3O4的优势相结合, 并且, 金属和碳材料的有机复合, 有望产生协同效应, 促进活化过程, 提高降解效率.
本研究从当前水污染治理面临的挑战出发, 利用高级氧化技术去除水中有机污染物高效、彻底的优势, 将氧化石墨烯独特的碳原子结构, 优异的电子传递能力和易于化学改性的特征相结合, 同时赋予材料磁性, 制备磁性氮掺石墨烯, 并对其进行多方位表征.然后以该新型金属碳基复合材料为催化剂, 过硫酸盐为氧化剂, 催化形成高活性的自由基, 用于污染水体中亚甲基蓝的降解.系统研究了降解中的主要影响因子, 催化反应动力学, 自由基生成情况以及亚甲基蓝的降解途径.该方法简单、高效, 有望为有机废水, 尤其是染料废水的处理提供一种简单、高效、无二次污染的技术方法支持.
2 材料与方法(Materials and methods)氧化石墨烯(GO)为多层粉末状, 购于苏州恒球石墨烯科技有限公司, 化学试剂亚甲基蓝、过硫酸钾、硫酸铁、硫酸亚铁、尿素、氢氧化钠、硫酸、氨水、甲醇、乙醇、KI、DMPO都为试剂纯, 购于国药集团化学试剂有限公司.实验全程用超纯水.所用的主要仪器设备包括:紫外-可见分光光度计(上海仪电分析仪器有限公司)、振动样品磁强计(VSM)(南京南大仪器)、JSM-6700F扫描电子显微镜(日本电子JEOL)、电子顺磁共振EPR (日本电子JES FA 200)、X射线衍射仪(德国布鲁D8)、BET比表面孔径分析仪(康塔Quantachrome, USA)、总有机碳分析仪(TOC-V CPH日本岛津).
2.1 磁性氮掺石墨烯的制备和表征称量0.5 g氧化石墨烯(GO)超声分散于150 mL水中.然后采用水热法共沉淀法将纳米四氧化三铁修饰到GO上面.具体操作为:往GO水溶液中分别加入一定量的硫酸亚铁和硫酸铁, 使亚铁和铁的物质的量的比为2:3, 在85 ℃的水浴恒温中搅拌溶解, 然后将机械搅拌速度调制500~600 r·min-1, 加入4 mol·L-1氨水, 调节体系pH为9~11, 继续搅拌45 min, 得到磁性GO(M-GO).将M-GO磁性分离清洗, 烘干研磨后, 再进行氮掺处理.以尿素为氮源和还原剂, 将制备的M-GO分散于水中, 加入一定量的尿素, 使尿素与GO的质量比为30:1, 充分溶解混合后, 将溶液体系置于180 ℃的水热反应釜中反应18 h, 得到磁性氮掺石墨烯M-N-G.对于氮掺石墨烯(N-G)的制备, 直接以GO而非M-GO为原料, 尿素为氮源和还原剂, 在180 ℃的水热反应釜中反应18 h得到.采用SEM、BET、XRD、VSM技术手段对其磁性氮掺石墨烯的表面形貌和结构、化学性质进行分析.
2.2 磁性氮掺石墨烯活化过硫酸酸钾体系的构建和参数优化往50 mL 10 mg·L-1的亚甲基蓝溶液中分别加入10 mg的M-N-G和0.4 mmol·L-1的过硫酸钾溶液, 水浴摇床避光处理一定时间, 考察其降解亚甲基蓝的效果.其中采用UV-VIS (紫外可见分光光度计)在665 nm下测定亚甲基蓝浓度.亚甲基蓝的降解率用Ct/C0表示, 其中Ct为t时刻溶液体系的亚甲基蓝的浓度, C0为亚甲基蓝的初始浓度.
考察溶液体系pH在2~8范围内, 对降解效果的影响.分别采用氢氧化钠和硫酸调节pH.考察处理时间和温度对降解效果的影响.分别在15~32 ℃内, 不同的时间点(5~120 min内取样), 测定降解效率, 对其进行降解动力学和热力学分析.另外对过硫酸钾的用量和M-N-G的投加量也进行了探究.降解亚甲基蓝过程中TOC的测定也是以50 mL, 10 mg·L-1亚甲基蓝为基准液体, 加入10 mg·L-1 M-N-G和0.4 mmol·L-1的过硫酸钾, 每隔一定时间测定体系中的TOC, TOC的去除率采用(TOC0-TOCt)/TOC0×100%公式来计算, 其中TOC0是10 mg·L-1亚甲基蓝的总有机碳含量, TOCt是t时刻测得的总有机碳含量.使用后的M-N-G, 先用0.1 mol·L-1的硫酸清洗, 然后再用超纯水清洗烘干重复利用.
3 实验结果与分析(Results and discussion) 3.1 材料的表征分析制备的M-N-G表观上为黑色的粉末状, 当其分散于水中, 在外部磁铁的作用下, 可以很好的分离.为了进一步了解材料的微观结构和性能, 首先采用场发射扫描电镜对GO、N-G和M-N-G进行了扫描.结果如图 1所示.
 |
| 图 1 GO(a)、N-G(b)M-N-G(c)的SEM图像 Fig. 1 SEM images of GO(a), N-G(b) and M-N-G(c) |
从图 1可以看出, GO为片层状结构, 是多层GO叠加在一起的结果, 表面不平整, 厚度大概在5~8 nm之间, 而经过磁性修饰和氮掺处理后, 材料的团聚性增加, 表面有大量Fe3O4颗粒沉积, 其中Fe3O4的粒径500 nm到1 μm不均匀分布, Fe3O4的粒径较大且分布比较宽, 可能是因为在掺杂氮的水热反应处理中, 沉积在氧化石墨烯上的Fe3O4进一步聚合而导致的.在SEM图像的基础上, 采用XRD, 进一步分析材料成分.结果如图 2所示.材料出现了Fe3O4的典型衍射峰.说明Fe3O4被很好的修饰到GO上面.振动样品磁强计分析的磁性强度为42.5 emu·g-1.而表观的结果也证实:材料具有良好的磁性, 在外部磁场的作用下, 分离效果良好, 如图 3所示.
 |
| 图 2 M-N-G的XRD图谱 Fig. 2 XRD pattern of M-N-G |
 |
| 图 3 M-N-G分散在水中的状态(a)及与外部磁铁作用的分离效果图(b) Fig. 3 The distribution status of M-N-G (a) and the magnetic separation of M-N-G (b)in aqueous solution |
对同一个样品分别进行了磁性分离和离心分离操作处理, 分离固液两相测定亚甲基蓝浓度, 发现两者的测量结果相差0.1%, 说明磁性分离操作非常有效.在后续的操作处理中, 测定样品前的分离操作都是在外部磁场作用下完成的, 避免离心操作处理的繁琐.
采用BET比表面积和孔径分析仪对合成的材料进行了分析.本研究中所用GO的比表面积约为175 m2·g-1(试剂公司提供), 修饰磁性Fe3O4后, 材料的比表面积降低到117.32 m2·g-1, 经过氮掺处理, 材料的比表面积进一步降低, 为94.35 m2·g-1.这也从另外一个方面说明磁性修饰和掺氮处理是发生在GO的表面的, 通过修饰Fe3O4和掺氮处理, 会使GO的比表面积降低.GO经过磁性修饰和氮掺处理, 其孔径有所降低.其孔径分别为5.221 nm(GO)、3.8605 nm(M-GO)、3.745 nm(M-N-G).根据IUPAC分类(Sing, 1989), 结合两幅图的特征判定, M-GO的氮气吸附脱附曲线是H4滞后环的IV型曲线, 而M-N-G的吸脱附曲线是具有H3滞后环的IV型曲线.结合GO本身的结构特征和滞后环的类型可以判定, 介孔的来源主要是片状GO堆积形成的狭缝孔.而经过修饰后, 孔径的降低, 其可能的原因是, 新引入的物质会进入到GO的夹缝结构, 使其孔径变小.
 |
| 图 4 M-GO (a)和M-N-G(b)的N2吸脱附曲线及BJH孔径分布图 Fig. 4 N2 adsorption-desorption curves and BJH pore distribution for M-GO (a) and M-N-G(b) |
为了选择最佳的材料作为催化剂, 分别调查了氮掺石墨烯(N-G, 5 mg)、磁性氧化石墨烯(M-GO, 10 mg)、磁性氮掺石墨烯(M-N-G, 10 mg)、Fe3O4(10 mg)作为吸附剂和催化剂, 以及单独加入过硫酸钾(0.4 mmol·L-1)处理亚甲基蓝的效果.从图 5可以看出, N-G的吸附效果最好, 这主要是因为该材料中石墨烯的比例最高.M-N-G的催化性能最好, 稍微优于M-GO的催化效果, 这是因为氮掺处理, 引入氮原子进入到GO的结构中, 一方面提高了GO的导电性, 另一方面增加了GO的缺陷结构, 更利于电子的传输(苏鹏等, 2012;王金双等, 2017).单独的Fe3O4的吸附和催化效果都不具备优势, 但和只加入过硫酸钾相比, 加入Fe3O4后, 降解率有所增大, 说明Fe3O4具备一定的催化效果, 该结果和之前赵进英研究报道一致(2010).综上比较, 在后续的实验中采用M-N-G作为催化剂.
 |
| 图 5 不同材料的吸附催化效果对比 Fig. 5 Adsorption and catalytic performances of different materials |
高级氧化技术运用于水处理的时候, 溶液体系的pH对降解效率影响显著.因为pH可以影响催化剂的活性, 氧化剂的氧化性能以及被处理污染物的存在状态.在本研究中, 首先研究体系pH对降解效率的影响.结果如图 6所示.
 |
| 图 6 pH值对亚甲基蓝降解效率的影响 Fig. 6 The effect of pH on the degradation of methylene blue |
从图中可以看出, 在pH=3~11范围内, 降解率都高于60%, 优于传统的芬顿氧化技术.这也和大部分文献报道相符.酸性pH的降解率(90%~95%)明显好于中性和碱性条件(62%~72%), 且酸性条件各个pH值的降解率差异不大.其可能的原因是:酸性条件下利于活化剂中Fe2+的释放, 而释放的Fe2+催化过硫酸钾形成硫酸根自由基, 用于亚甲基蓝的降解.随着pH的升高, Fe2+的释放减缓, 硫酸根自由基产生减少, 并且碱性条件下利于羟基自由基的形成, 相比于硫酸根自由基(t1/2=30 ~40 μs), 羟基自由基(t1/2 < 1 μs)的寿命更短(Oh et al., 2016), 被有效利用的自由基数量降低.本研究中, 亚甲基蓝自身溶液的pH接近6, 体系在该溶液条件下, 可以保持一个较高的降解效率, 因此, 后续处理中, 没有对体系的pH值进行调节.
3.4 处理温度对降解的影响降解速率常数是氧化降解技术中的一个重要参数, 直接关系到该技术方法的效率和成本.其中温度对催化降解反应的速率常数影响显著.在本研究中, 探究15、21、32 ℃下, 材料的催化性能.考虑材料具有较大的比表面积, 对亚甲基蓝有一定的吸附作用.首先将M-N-G加入到亚甲基蓝溶液中, 吸附处理1 h后, 再加入过硫酸钾进行催化降解处理, 每隔一定时间取样测定浓度.结果如图 7所示.
 |
| 图 7 温度对亚甲基蓝去除率的影响 Fig. 7 The effect of temperature on the degradation of methylene blue |
从图 7可以看出, M-N-G对亚甲基蓝有一定的吸附效果, 在15~32 ℃的条件下, 吸附可以去除体系中15%~25%的亚甲基蓝.加入过硫酸钾以后, 亚甲基蓝的去除率显著提高, 2 h以后, 降解率约为93%.随着温度的升高, 降解速度增加, 在2 h的催化反应过程中, 前1 h反应较快, 可认为是催化反应的快速反应阶段, 第2 h可认为是慢速反应阶段.采用伪一级动力学方程进行拟合, 发现15、21、32 ℃时对应的降解速率常数k分别为0.0227、0.0271、0.0488 min-1.根据阿伦尼乌斯公式(lnk=lnA-Ea/RT, lnk~1/T作图, 由斜率可得表观活化能Ea)计算的活化能为33.7 kJ·mol-1.Wen等(2014)使用零价铁活化过硫酸盐降解对硝基氯苯的活化能为31.79 kJ·mol-1, Kang等(2016)使用氮掺杂还原氧化石墨烯活化过硫酸盐降解抗生素的活化能为38.6 kJ·mol-1.
3.5 催化剂和过硫酸钾初始浓度对降解效率的影响考察了加入不同浓度催化剂, 对亚甲基蓝降解效率的影响.从图 8a可以看出, 0.5 mmol·L-1过硫酸钾可以去除体系中约40%的亚甲基蓝.这是因为过硫酸钾作为一种氧化剂, 其标准氧化还原电位E0=1.96 V, 因此可以在一定程度上降解亚甲基蓝.催化剂的加入, 显著提高了污染物的去除率.随着体系中活化剂的用量从80 mg·L-1增加到280 mg·L-1, 降解效率从70%增加到了95%.其中200 ~280 mg·L-1的情况下, 降解效率几乎接近, 这些说明尽管活化剂用量对体系降解效率影响重大, 但当增加到一定浓度的时候, 这种影响基本消失.其原因是, 体系中过硫酸钾的量是有限的, 当M-N-G增加到200 mg·L-1时, 以能够满足催化0.5 mmo·L-1所需要的量, 因此, 再增加活化剂的量影响不大.
 |
| 图 8 M-N-G用量(a)及过硫酸钾用量(b)对降解效率的影响 Fig. 8 The effect of catalyst dosage (a) and oxidant dosage (b) on degradation of methylene blue |
在分析M-N-G加入量对降解效率的影响后, 分析了氧化剂过硫酸钾用量的影响.从图 8b可以看出, M-N-G具备一定的吸附能力, 吸附去除率约为20%.因为M-N-G的比表面积为94.35 m2·g-1(BET分析结果), 可以通过表面吸附去除溶液中的亚甲基蓝, 并且石墨烯上面的π键也可和亚甲基蓝作用进行吸附.当过硫酸钾的浓度从0.02 mmol·L-1增加到0.5 mmol·L-1时, 降解效率从50%增加到了95%, 并且从0.2 mmol·L-1到0.5 mmol·L-1, 降解效率都在90%以上, 变化不大.这些说明, 过硫酸钾经过活化后, 形成自由基, 降解亚甲蓝的能力显著提高.而当过硫酸钾浓度增大到一定程度的时候, 继续加大用量, 效果微小, 这是因为体系中的M-N-G的量是一定的, 只能有效活化一定量的过硫酸钾, 并且随着反应的进行, 中间产物的生成, 活化位点的部分失活等因素, 也会导致活化剂的活性降低.尽管已有研究表明过硫酸盐可以作为淬灭剂与硫酸根自由基反应(Anipsitakis et al., 2003), 但在图 8b所示的结果中, 并没有显著发生.其可能原因是:发生自身漼灭反应的速度比氧化降解亚甲基蓝的速度小;并且M-N-G具备一定的吸附亚甲基蓝的溶液, 而催化形成硫酸根自由基优先分布在M-N-G的周围, 利于硫酸根自由基和亚甲基蓝快速相互作用, 因此在本体系中, 较多的过硫酸钾并没有对体系产生抑制作用.
图 8优化了体系催化剂和氧化剂的用量, 以得到最佳的降解效果.通过优化:当体系氧化剂浓度为200 mg·L-1, 过硫酸钾浓度为0.4 ~0.5 mmol·L-1, 对10 mg·L-1的亚甲基蓝的去除率高达95%, 但是矿化程度如何?图 9为体系在不同条件下的矿化率.可以看出, 最高约为50%, 这说明亚甲基蓝在降级的过程中, 除开部分矿化为CO2和H2O外, 还有其他有机物质的生成.
 |
| 图 9 亚甲基蓝去除过程中的总有机碳TOC的去除率 Fig. 9 The removal efficiency of TOC during the degradation methylene blue |
已有研究结果表明:当过硫酸盐被活化后, 能够产生硫酸根自由基, 然后硫酸根自由基又可以和H2O或OH-反应, 生成羟基自由基.通常体系通过硫酸根自由基和羟基自由基的作用去除污染物(Fang et al., 2013; 2017).当用M-N-G活化过硫酸钾的时候, 体系中自由基的生成情况如何?通过EPR分析技术和自由基漼灭实验可以一探究竟.采用0.1 mol·L-1 DMPO作为捕获剂, 调节体系pH为6, 在室温下的测定结果如图 10所示.硫酸根自由基和羟基自由基的信号都有检测到.其中•代表羟基自由基的四重峰(1:2:2:1), ▲代表硫酸根自由基的六重峰(1:1:1:1:1:1), 说明体系中两种自由基共同发挥作用.甲醇和乙醇通常被用作化学淬灭剂来抑制羟基自由基和硫酸根自由基的作用.硫酸根自由基与甲醇和乙醇反应的速率常数分别为3.2×106、1.6×107 L·mol-1·s-1, 而羟基自由基与甲醇和乙醇反应的速率常数分别为:9.7×108、1.9×109 L·mol-1·s-1(Oh et al., 2016), 考察了甲醇和乙醇的加入对降解效率的影响(分别使用10和100倍过硫酸钾浓度).结果如图 11所示:两种醇对降解效率影响微小, 而加入KI后(1倍和2倍过硫酸钾浓度), 降解效率显著降低.甲醇和乙醇作为常用的淬灭剂, 在本实验中, 没发挥作用, 而KI的漼灭效果良好.类似的现象也被报道(Xu et al., 2012; Wang et al., 2015).其可能的原因是因为亲水性的醇类物质分散在水中, 不容易与形成在M-N-G表面的自由基反应.
 |
| 图 10 催化反应系统的EPR图谱 Fig. 10 The EPR image of this catalytic system using DMPO as trapping agent |
 |
| 图 11 不同淬灭剂下的降解效果 Fig. 11 The effect of on degradation of methylene using methanol, ethanol and KI as scavengers |
以上研究结果表明:亚甲基蓝的去除以自由基氧化降解为主, 同时也有部分来自于过硫酸钾的直接氧化, 以及M-N-G的吸附去除.而M-N-G活化过硫酸钾是通过Fe2+被氧化成Fe3+提供电子, 而GO作为电子载体, 达到高效催化过硫酸钾产生硫酸根自由基.
3.7 催化剂重复利用性M-N-G作为催化剂, 其重复利用次数对材料的综合性能评价至关重要.使用后的活化剂, 在外部磁场的作用下, 从体系中分离出来, 然后采用0.1 mol·L-1 H2SO4清洗后, 再用超纯水清洗, 重复利用4次的结果如图 12所示.
 |
| 图 12 活化剂重复利用效果图 Fig. 12 The recycle capability of M-N-G catalyst |
从图中可以看出, 第4次使用时, 其降解率依然可达60%, 具备较好的重复利用效果.导致催化剂活性变差的原因可能是在使用中, 活性位点被吸附在催化剂表面的亚甲基蓝及中间产物占据, 导致其减少, 另外反复使用的过程中, 活性位点会逐渐失活.
4 结论(Conclusions)本研究中探讨了使用M-N-G作为活化剂, 催化过硫酸钾产生硫酸根自由基降解水中亚甲基蓝的效果.所述M-N-G的制备方法简单, 磁性良好, BET比表面积为94.35 m2·g-1, 孔道大小为3.745 nm.体系在pH 2~6时, 能够降解去除体系中90%的亚甲基蓝, 而中性和碱性条件下, 降解效率低于70%.通过降解动力学和热力学分析表明:在15~32 ℃下, 体系的降解速率常数在0.042 min-1到0.109 min-1之间, 反应的活化能为33.7 kJ·mol-1.通催化剂和氧化剂用量优化研究表明:M-N-G具备有限的吸附去除亚甲基蓝的能力, 过硫酸钾也可以通过自身的氧化能力降解部分亚甲基蓝.而当磁性氮掺GO的浓度为200 mg·L-1, 过硫酸钾浓度为0.2~0.5 mmol·L-1时, 能够降解去除95%浓度为10 mg·L-1的亚甲基蓝, 且TOC的去除率可达50%.EPR分析结合自由基漼灭实验结果表明:体系中主要以生成硫酸根自由基为主.重复实验结果表明:该催化剂可以反复利用4次后, 降解效率依然可达60%.本研究所展示的技术方法简单、高效, 操作方便, 为污染水体的有机污染物治理, 尤其是染料废水治理提供理论依据和技术借鉴.
Anipsitakis G P, Dionysiou D D. 2003. Degradation of organic contaminants in water with sulfate radicals generated by the conjunction of peroxymonosulfate with cobalt[J]. Environmental Science & Technology, 37(20): 4790–4797.
|
Duan X, Sun H, Kang J, et al. 2015. Insights into heterogeneous catalysis of persulfate activation on dimensional-structured nanocarbons[J]. Acs Catalysis, 5: 4629–4636.
DOI:10.1021/acscatal.5b00774
|
Fang G D, Gao J, Dionysiou D D, et al. 2013. Activation of persulfate by quinones: free radical reactions and implication for the degradation of PCBs[J]. Environmental Science & Technology, 47(9): 4605–4611.
|
Fang G D, Wu W H, Liu C, et al. 2017. Activation of persulfate with vanadium species for PCBs degradation: A mechanistic study[J]. Applied catalysis B: Environmental, 202: 1–11.
DOI:10.1016/j.apcatb.2016.09.006
|
Ghauch A, Tuqan A M, Kibbi N, et al. 2012. Methylene blue discoloration by heated persulfate in aqueous solution[J]. Chemical Engineering Journal, 213(1): 259–271.
|
Kang J, Duan X G, Zhou L, et al. 2016. Carbocatalytic activation of persulfate for removal of antibiotics in water solutions[J]. Chemical Engineering Journal, 288: 399–405.
DOI:10.1016/j.cej.2015.12.040
|
Lee H, Lee H J, Jeong J, et al. 2015. Activation of persulfates by carbon nanotubes: Oxidation of organic compounds by nonradical mechanism[J]. Chemical Engineering Journal, 266: 28–33.
DOI:10.1016/j.cej.2014.12.065
|
李湘, 李忠, 罗灵爱. 2006. 活性炭吸附二苯并呋喃的动力学[J]. 环境科学学报, 2006, 26(10): 1695–1700.
DOI:10.3321/j.issn:0253-2468.2006.10.019 |
刘元坤, 王建龙. 2017. 磺胺二甲基嘧啶在过硫酸盐存在下的辐照降解研究[J]. 环境科学学报, 2017, 37(2): 652–655.
|
马龙, 王雅洁, 杨成. 2016. 废水高级氧化技术研究现状与发展[J]. 环境工程, 2016, 34(6): 52–55.
|
Mielke, Paul W. 2009. A novel advanced oxidation process to degrade organic pollutants in wastewater: Microwave-activated persulfate oxidation[J]. Journal of Environmental Sciences, 21(9): 1175–1180.
DOI:10.1016/S1001-0742(08)62399-2
|
Ocampo A M. 2009. Persulfate activation by organic compounds [D]. Washington:Washington State University
http://search.proquest.com/docview/305019336 |
Oh W D, Dong Z L, Lim T T. 2016. Generation of sulfate radical through heterogeneous catalysis for organic contaminants removal: Current development, challenges and prospects, Applied Catalysis B: Environmental[J]. Applied Catalysis B Environmental, 194: 169–201.
DOI:10.1016/j.apcatb.2016.04.003
|
Oh W D, Lu S K, Dong Z L, et al. 2015. Performance of magnetic activated carbon composite as peroxymonosulfate activator and regenerable adsorbent via sulfate radical-mediated oxidation processes[J]. Journal of Hazardous Materials, 284: 1–9.
DOI:10.1016/j.jhazmat.2014.10.042
|
Oturan M A, Aaron J J. 2014. Advanced oxidation processes in water/wastewater treatment: principles and applications a Review[J]. Critical Reviews in Environmental Science and Technology, 44(23): 2577–2641.
DOI:10.1080/10643389.2013.829765
|
欧阳磊, 丁耀彬, 朱丽华, 等. 2013. 钴掺杂铁酸铋活化过硫酸盐降解水中四溴双酚A的研究[J]. 环境科学, 2013, 34(9): 3508–3511.
|
Pu M, Ma Y, Wan J, et al. 2014. Fe/S doped granular activated carbon as a highly active heterogeneous persulfate catalyst toward the degradation of Orange G and diethyl phthalate[J]. Journal of Colloid & Interface Science, 418: 330–337.
|
庞娅. 2012. 磁性纳米复合材料的制备及其去除水体污染物的研究[D]. 长沙: 湖南大学
http://cdmd.cnki.com.cn/Article/CDMD-10532-1012482437.htm |
Sing K S W. 1989. The Use of Gas Adsorption for the Characterization of Porous[J]. Solids Colloids and Surfaces, 38: 113–124.
DOI:10.1016/0166-6622(89)80148-9
|
苏鹏, 郭慧林, 彭三, 等. 2012. 氮掺杂石墨烯的制备及其超级电容性能[J]. 物理化学学报, 2012, 28(11): 2745–2753.
DOI:10.3866/PKU.WHXB201208221 |
Wang X B, Qin Y L, Zhu L H, et al. 2015. Nitrogen-doped reduced graphene oxide as a bifunctional material for removing bisphenols: synergistic effect between adsorption and catalysis[J]. Environmental Science Technology, 49: 6855–6864.
DOI:10.1021/acs.est.5b01059
|
王金双, 秦川丽, 王虹涧, 等. 2017. 氮掺杂石墨烯与g-C3N4二维异质体对纳米Fe2O3光催化剂的复合改性[J]. 高等学校化学学报, 2017, 2(38): 246–251.
|
Wen Q, Zhang H, Xu H B, et al. 2014. Degradation of p-nitrochlorobenzene in soil by zero-valent iron activated persulfate[J]. Journal of Nanjing Agricultural University, 6: 1230–1235.
|
Xu L J, Wang J L. 2012. Magnetic nanoscaled Fe3O4/CeO2 composite as an efficient Fenton-like heterogeneous catalyst for degradation of 4-chlorophenol[J]. Environmental Science Technology, 46: 10145–10153.
DOI:10.1021/es300303f
|
肖蓝, 王祎龙, 唐玉霖. 2013. 石墨烯及其复合材料在水处理中的应用[J]. 化学进展, 2013, 25(Z1): 419–430.
|
Zhang N, Qiu H, Liu Y, et al. 2011. Fabrication of gold nanoparticle/graphene oxide nanocomposites and their excellent catalytic performance[J]. Journal of Materials Chemistry, 21(30): 11080–11083.
DOI:10.1039/c1jm12539g
|
张琼, 贺蕴秋, 陈小刚. 2010. 氧化钛/氧化石墨烯复合结构及其光催化性能[J]. 科学通报, 2010, 55(7): 620–628.
|
张瑛洁, 马军, 宋磊, 等. 2009. 树脂负载Fe/Cu多相类芬顿降解染料橙黄[J]. 环境科学学报, 2009, 29(7): 1419–1425.
|
赵艳丽. 2016. 三维氮、硫掺杂石墨烯材料的制备及其在染料敏化太阳能电池中的应用研究[D]. 哈尔滨: 哈尔滨工业大学
http://cdmd.cnki.com.cn/Article/CDMD-10213-1016774748.htm |
赵进英. 2010. 零价铁/过硫酸钠体系产生硫酸根自由基氧化降解氯酚的研究[D]. 大连: 大连理工大学
http://cdmd.cnki.com.cn/Article/CDMD-10141-2010111263.htm |