 2017, Vol. 37
2017, Vol. 37



芳香烃是由苯环构成的一类碳氢化合物.自然背景下, 森林火灾和火山喷发等是芳香烃最主要的来源;随着现代工业的发展, 煤和石油等化石燃料的大量使用已引起环境中芳香烃急剧增加, 造成了一系列的环境污染问题.据报道, 污染土壤中芳香烃含量已从μg·kg-1上升到mg·kg-1量级, 检出率从不到20%上升到80%以上 (赵其国等, 2009;Zhang et al., 2009);我国长江口滨岸潮滩表层沉积物中芳香烃总量分布范围在0.263~6.372 mg·kg-1之间 (刘敏等, 2001);北方老工业地区空气中全年芳香烃总含量变化范围为16.3~712.1 ng·m-3(李伟芳等, 2013).由于芳香烃化合物的疏水亲脂性, 它们很容易被生物体吸收并累积, 与细胞内的DNA内碱基结合, 引起基因功能的损伤, 产生致癌性、致畸性和致突变性的“三致”基因污染效应, 严重危害人群健康和生态安全.
芳香烃类污染物对基因造成的损伤污染与它们之间的结合作用密切相关, 因此, 理解这些化合物对基因污染的前提是必须首先阐明“污染物-基因碱基”间相互作用规律.通常的观点认为, 进入细胞内的芳香烃可被胞内酶催化氧化并进一步的羟基化, 产生以芳烃为母体结构的羟基芳香烃 (Hu et al., 2015).由于羟基芳烃拥有和基因碱基类似的结构, 在基因复制的过程中细胞使用了羟基芳烃原料, 产生了错误的基因编码方式, 引起基因的污染损害, 导致“三致”效应.同时, 本课题组最近的研究发现, 芳香烃类污染物也可以不经过酶催化作用直接对DNA碱基产生影响, 干扰基因的转化和表达, 表现基因毒性 (Kang et al., 2013;Kang et al., 2015).这些研究表明, 无论是酶催化还是物理影响, 芳香烃类污染物与基因碱基最初的非共价结合是产生基因毒性的基础.因此, 要探明芳烃污染物对基因的污染特征, 必须首先阐明它们之间的相互作用规律.然而迄今为止, 关于二者间的弱相互作用及驱动机制, 国内外相关报道仍旧很少.
目前, 针对使用吸附方法探究部分可溶小分子吸附剂-吸附质间相互作用规律的研究存在3个制约因素:第一, 因为基因碱基分子在水溶液中存在一定的溶解性, 所以基于传统离心分离吸附剂的方法无法探究这种基于分子对分子的吸附过程 (Hu et al., 2008);第二, 吸附剂-吸附质间相互作用力的定量化研究仍有待开拓 (Kang et al., 2015);第三, 吸附剂-吸附质间相互作用主要依赖于非化学成键的弱作用力, 因此,如何精确地甄别非共价弱相互的作用力类型成为又一个制约因素 (Johnson et al., 2010).针对这3个问题, 本研究拟使用等温吸附-光纤固相萃取和定量-构效关系的方法探究基因碱基-芳香烃相互作用规律, 并利用红外光谱法 (FTIR)和能量计算找出基因碱基上的结合位点.以期系统地阐释芳香烃菲模型污染物与基因碱基之间的弱相互作用及驱动机制, 试图为深入揭示芳香烃对基因污染的原理提供理论基础.
2 材料与方法 (Materials and methods) 2.1 试剂菲购于西格玛奥德里奇有限公司 (Sigma-Aldrich, 美国), 纯度≥97.0%, 相对分子质量为178.24, 25 ℃纯水中溶解度为1.18 mg·L-1, 辛醇-水分配系数 (lgKow) 为4.46.腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧啶纯度高于98.0%, 购自沃凯生物试剂有限公司 (上海, 中国).固相微萃取使用的光纤为美国莫仕 (Molex) 有限公司生产的聚二甲基硅氧烷涂层石英光导纤维 (内芯110 μm, 涂层30 μm).助溶剂甲醇为色谱纯 (99.0%), 购于美国天地试剂有限公司.溶剂水为美国密理博 (Millipore) 超纯水仪 (Milli-Q) 生产的超纯水.
2.2 等温吸附-光纤萃取实验等温吸附和光纤微萃取的联合方法被用来研究基因碱基与菲之间的结合作用, 该方法已被证明针对芳香烃的萃取回收率高达98.9%(Hu et al., 2008).称取0.01 mg碱基置于20 mL的透明螺旋口玻璃瓶内 (聚四氟乙烯垫片), 称重并记录;加超纯水20 mL, 分别使用0.05 mol·L-1的NaOH或HNO3水溶液调节pH值至5.00、7.00和9.00(±0.05), 再次称重并记录;2次称重的差值为水溶液质量, 除以配制溶液温度下的水密度可得到水溶液体积.加入菲母液以获取6组所需要的菲初始浓度, 加入母液的体积不高于瓶内溶液总体积的0.1%, 以减小甲醇溶剂对吸附平衡产生的误差.加入长度为1 cm的石英光纤至瓶内, 并使用内衬垫为聚四氟乙烯材质的瓶盖密封瓶口.所有样品置于25 ℃恒温的旋转培养器中平衡24 h后, 使用不锈钢镊子取出石英光纤, 移置于容积为1 mL的液相棕色小瓶中, 加入250 μL甲醇解析24 h后移除光纤.使用液相色谱法测定萃取至甲醇中的菲浓度.以上每个浓度梯度设3个平行组, 文中展示的结果为均值.标准曲线至少设置8个标准点, 除未添加吸附剂和吸附质之外, 其它操作与吸附-固相萃取实验过程相同.
2.3 测定方法使用高效液相色谱法 (HPLC, LC-20A, 岛津, 日本) 分析溶液中的菲浓度.色谱柱为岛津Inertsil ODS-P液相柱 (规格为5 μm, 250 × 4.6 mm I.D.), 流动相为甲醇 (色谱纯), 流速l.0 mL·min-1, 柱温40 ℃, 进样量20 μL, 检测波长为254 nm (SPD-20A, 岛津).
2.4 红外光谱红外光谱 (FTIR) 法被用来定性地分析碱基官能团变化.吸附平衡后, 溶液在5000 g下离心10 min, 移除未沉降的上液体.沉淀样品在-65 ℃下真空干燥48 h, 获得粉末状样品.该离心分离操作仅被用来获取碱基, 少量的碱基溶解损失不会对后续FTIR分析结果产生影响. KBr (200 mg, 光谱纯) 与碱基样品 (2 mg) 混合, 在6.0 t的压力下持续5 min制得1.3 cm直径的样片, 经傅里叶变换红外光谱仪 (TENSOR, 布鲁克, 德国) 分析获得光谱曲线.每克碱基中菲占碱基的质量比不超过3%, 不会对碱基的红外光谱产生干扰.
2.5 计算化学使用Gabedit软件 (Version 2.4.5, 美国)(Allouche, 2011) 构建了包括腺嘌呤、鸟嘌呤、胞嘧啶、胸腺嘧啶和菲在内的5个分子模型.4种碱基与菲通过AutoDock软件 (美国) 分别进行分子对接 (Trott et al., 2010), 确定分子对接点.所有模型首先通过半经验法 (Semi-Empirical Method PM7, Mopac2012, USA) 进行结构优化 (Stewart et al., 2012), 使用高斯计算软件 (Gaussian 09E0.1, 美国) 在B3LYP/6-31G (d, p) 计算水平对模型进行最终的优化和频率分析, 同时在DFT-D3(BJ) 水平对计算进行误差矫正 (Frisch et al., 2009).所有计算中, 使用隐式水溶剂模型.针对腺嘌呤、鸟嘌呤、胞嘧啶、胸腺嘧啶与菲的计算结果, 使用Gaussian view软件 (Dennington et al., 2009) 分析了它们的表面静电势, 分析方法及理论基础见参考文献 (Scrocco et al., 1973;Murray et al., 2011;Kang et al., 2016).
2.6 弱作用引力分析计算的结果通过波函数分析软件Multiwfn进一步分析 (Lu et al., 2012), 以获取“碱基-菲”相互作用力的等值面, 甄别作用力类型.成键类型可以通过以下的量子力学的电子密度法进行甄别 (Kang, 2015;Johnson et al., 2010):
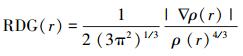
|
(1) |
式中, RGD (r) 为约化密度梯度;ρ(r)、|▽ρ(r)|和分别为量子力学电子密度、梯度算子和子力学电子密度的梯度算子模运算.分析碱基与菲之间的成键类型后, 在2个分子间使用带不同色彩的等值面呈现非共价键类型.绿色和红色分别代表分子间非共价引力作用和分子内部斥力.当特征值Sign (λ2)ρ > 0时, 表示对应的分子内部的斥力;当该特征值≤0时, 表示弱的分子力作用.基于负性强弱, 区分非共价作用键级.
3 结果与讨论 (Results and discussion) 3.1 基于静电引力的碱基-菲弱相互作用图 1分别显示的是腺嘌呤、鸟嘌呤、胞嘧啶、胸腺嘧啶和菲分子的表面静电势.红色部分代表负电势占据区域, 蓝色代表正电势占据区域, 颜色越深, 电势越高.腺嘌呤母体是由2个具冗余电子的CN环构成, 即CN 6环和CN 5环结构.从腺嘌呤的表面静电势图 (图 1a) 可以看出, 蓝色正电势位于母体上的氢 (—H)和氨基 (—NH2) 取代基上, 而环状结构母体表面整体电势较低, 为白色或橙红色低电势区域.图 1b为鸟嘌呤的表面静电势图, 最具明显的特征是负电势位于羰基 (O) 取代基 (红色), 中等电势区域位于母体结构平面上 (白色), 正电势位于母体取代基—NH2和—H上.
 |
| 图 1 腺嘌呤 (a)、鸟嘌呤 (b)、胞嘧啶 (c)、胸腺嘧啶 (d)4种基因碱基和菲 (e) 的表面静电势图 (ESP) Fig. 1 Electrostatic potential of four genetic bases, including adenine (a), guanine (b), cytosine (c), and thymine (d), and phenanthrene (e) |
图 1c为胞嘧啶分子的表面静电势.相比于嘌呤结构, 胞嘧啶仅由一个不饱和电子的CN 6环结构构成, 取代基为一个氨基 (—NH2)和一个羰基 (O).从图中可以看出, 取代基羰基及母体N为表面红色负电势所覆盖.类似地, 胸腺嘧啶母体结构 (图 1d) 上有2个间位羰基 (O), 均呈现红色的表面负电势.同时, 位于母体结构上的取代基氢 (—H)和甲基 (—CH3) 表面均为蓝色, 为正电势所覆盖的区域.可知, 无论是嘌呤还是嘧啶结构, 氧官能结构均为负电势表面, 而N、C和H结构表面均呈现正电势.
吸附质菲分子的表面静电势可以从图 1e观察到.菲分子的外表面均为淡蓝色静电势所覆盖, 而芳香核的内部区域均为淡红色.该结果表明, 菲的表面负电势集中于芳香核内部的电子富集区域, 而表面正电势分布于分子外围.因为菲分子是平面结构, 所以无论它的正电势还是负电势表面均可暴露于外围环境中与其它分子通过静电引力相互作用.先前研究也表明, 由于芳香核为负电子富集的不饱和区域, 能够通过π-π或π-阳离子等方式与其它化合物或吸电子基团 (阳离子) 相互作用 (Chen et al., 2007;Zhu et al., 2005).这也是芳香烃污染物与吸附剂之间相互作用的最主要作用力类型之一.同时, 值得注意的是, 4种碱基母体结构上电势均为相对的贫电子区域, 而—H和—NH2均为正电势区域;中心区域为负电势的菲可能更倾向于与这些贫电子或正电势区域发生π-π或π-吸电子静电引力作用.
分子间引力作用的本质是原子或基团外围电子之间的相互作用, 因此,可以基于分子表面静电势和吸附实验来判定碱基-菲相互作用规律.4种基因碱基对芳香烃菲的等温吸附 (25 ℃) 可通过Freundlich吸附模型描述:
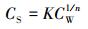
|
(2) |
式中, CS是固相吸附平衡浓度 (nmol·g-1);CW是吸附平衡后水相浓度 (nmol·L-1);1/n是线性指数, 无量纲;K是亲和系数 (nmol1-n·Ln·kg-1).图 2a所示为CS对CW绘制的等温吸附曲线.鸟嘌呤、胞嘧啶和胸腺嘧啶对菲的吸附呈现线性吸附样式, 表明吸附质菲在水-吸附剂间发生分配作用;而腺嘌呤的吸附呈现下凸的形式, 表明吸附剂与菲间的引力非常弱 (近藤精一等, 2006).对Freundlich吸附模型进一步作线性变换, 可获得相关吸附参数:
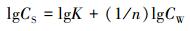
|
(3) |
 |
| 图 2 4种基因碱基对菲的吸附等温线 (25 ℃, pH=7.0) Fig. 2 Adsorption isotherm of phenanthrene on four gene bases (AGCT) at 25 ℃ and pH=7.0 |
lgCS对lgCW做图, 获得图 2b.腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧啶4种碱基对芳香烃菲的线性指数 (1/n) 分别是1.64、0.92、0.96和1.15.腺嘌呤对菲的吸附中, 线性指数1/n大于1.0, 再次证明它们之间的相互作用力较弱 (近藤精一等, 2006).值得注意的是, 包括鸟嘌呤、胞嘧啶和胸腺嘧啶在内的3种基因碱基对菲的线性指数约等于1.0, 均可被归结为吸附质在碱基分子和溶剂水间的分配效应.参照图 1关于静电势分析的结果, 除腺嘌呤结构外, 包括鸟嘌呤、胞嘧啶、胸腺嘧啶在内的3种碱基含有未饱和电子的氧取代基.这表明未饱和电子取代基可能增加分子结构电子未饱和量, 引起碱基母体π电子量的增加, 从而增强碱基-菲间基于π-π或π-吸电子的静电引力作用.
3.2 结合位点分子间引力产生的本质是电子之间的相互作用行为, 而电子相互作用能影响分子官能团振动的变化, 官能团振动变化和电子间相互行为可通过红外光谱和计算化学的结果进行表征.本文中腺嘌呤、鸟嘌呤、胞嘧啶、胸腺嘧啶及菲官能团的变化通过红外光谱法判定.由图 3可知, 腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧啶中含氮官能团的特征吸收谱带分别为1605、1710、1465和1662 cm-1(Taillandier et al., 1992;Loprete et al., 1993;Ouameur et al., 2004;Andrushchenko et al., 2002).对于腺嘌呤 (图 3a), 波数位于1675、1510和1460 cm-1的峰分别代表了腺嘌呤碱基上的—NH2、CC/CN伸缩振动、—CH2变角振动;在1372、1286和1245 cm-1上的3个吸收峰, 则表明醣—CH和醣2′OH变角振动的耦合 (虞宝珠等, 1986).由于结构类似性, 鸟嘌呤 (图 3b) 与腺嘌呤的母体具类似的光谱特征, 前者在1680 cm-1的峰可归属为取代基CO的伸缩振动.胞嘧啶比胸腺嘧啶少一个羰基 (O), 两者在1600 cm-1以下的峰相近, 表明这些峰与母体结构上CC/CN、—CH3和—CH的不同振动类型有关;而在1675 cm-1处, 胞嘧啶出现一个最大的峰, 可与CN6环母体上的羰基的振动相联系.相较于胞嘧啶, 胸腺嘧啶在1745 cm-1附近出现了一个峰, 表明另外一个羰基的存在.
 |
| 图 3 碱基-菲红外光谱图 (a.腺嘌呤, b.鸟嘌呤, c.胞嘧啶, d.胸腺嘧啶, 箭头所示为CN母体特征吸收峰) Fig. 3 FTIR of genetic bases associated with phenanthrene (a.adenine, b.guanine, c.cytosine, d.thymine, arrows represent the characteristic absorption peaks in CN rings) |
与菲结合后, 腺嘌呤分子上位于1605和1510 cm-1的峰变强, 而1460 cm-1处的峰减弱, 表明腺嘌呤母体上CC/CN参与到与菲的结合过程中.同时, 结合菲后, 腺嘌呤上位于1305、1330和1120 cm-1处出现新的光谱带, 可能与CN 5环上醣—CH和醣2′OH的变化有关.无论光谱峰是增强还是减弱, 这些变化均表明腺嘌呤母体结构上CN 6环和5环结构均参与到与菲的结合过程中.鸟嘌呤结合菲后 (图 3b) 位于1710 cm-1的特征峰变弱, 表明CN母体结构也参与到对菲的结合过程中.类似的结论也能从胞嘧啶 (图 3c) 的光谱变化中获得, 发现胞嘧啶结合菲后母体特征谱变弱 (1465 cm-1).这些结论均证明与菲结合后所有碱基母体结构均出现变化, 表明碱基以一种平面对平面的方式与菲结合.
图 4区分了碱基-菲间非共价弱相互作用的类型, 其中, 红色等值面表示分子内部斥力, 绿色表示分子间引力的存在.从图 4a可以看出, 腺嘌呤与菲的分子内部均观察到红色等值面, 表明分子内部斥力的存在.在腺嘌呤-菲分子间, 出现了2对绿色环状等值面, 分别正置于CN6环-苯核和CN5环-苯核间.结合图 1的分子静电势图, 在腺嘌呤分子内部存在2组以CN6环和5环结构的π电子, 同时菲芳香核内部存在3组环状结构的π电子, 所以腺嘌呤-菲间可通过上述电子发生π-π引力作用.该结果进一步证实了红外光谱的分析结果, 即碱基以一种平面对平面的方式与菲结合.图 4b为腺嘌呤-菲间作用力的定量化分析结果.横坐标特征值 (Sign (λ2)ρ) 可被界定为2个对应区间, 即 > 0和≤0的区间.当特征值 > 0时, 所揭示的是分子的内斥力;当特征值≤0时, 所呈现的是分子间引力.值负性越大, 引力越强.本文关注分子间引力, 即≤0的区间.从图中可以观察到, 在特征值0~0.02 a.u区间存在2个最为明显的特征值:零附近的特征值可与芳香核-CN 5环间的π-π引力相联系, 而-0.014 a.u特征值可被归结为芳香核-CN6环间的π-π引力.显然, 由于更多的π电子, 腺嘌呤CN6环对芳香核的引力强于与CN5环.
 |
| 图 4 基因碱基-芳烃菲之间的弱相互作用 (a, b.腺嘌呤-菲, c, d.鸟嘌呤-菲, e, f.胞嘧啶-菲, g, h.胸腺嘧啶-菲) Fig. 4 Weak interaction between genetic bases and phenanthrene (a, b.adenine-phenanthrene, c, d.guanine-phenanthrene, e, f.cytosine-phenanthrene, g, h. thymine-phenanthrene) |
尽管鸟嘌呤比腺嘌呤多一个羰基, 但两者与菲的引力类型一致.从图 4c可看到, 在鸟嘌呤-菲间出现了2组蓝色的环状等值面, 分别对应于CN5环-苯核和CN6环-苯核, 这表明鸟嘌呤-菲间也存在π-π引力.图 4d显示了引力特征值的分析结果, 除了对应于红色等值面的分子内部的斥力外 (Sign (λ2)ρ > 0), 在特征值0~-0.002 a.u间出现2个重要特征值带 (-0.002与-0.015 a.u), 分别与芳香核-CN 5环和芳香核-CN6环间π-π引力有关.该结果进一步表明鸟嘌呤-菲间引力依赖于π-π引力.
图 4e为胞嘧啶-菲间引力等值面图.如图所示, 胞嘧啶-菲间仅存在一个绿色等值面, 且胞嘧啶的几何中心正置于菲弧角位 (粉红色箭头) 碳原子.图 1e表明菲弧角位碳原子均为相对寡电子区域, 因此, 该位碳原子与嘧啶上的CN6环富电子区域可通过“吸电子-赠电子”静电引力相互吸引.定量化分析结果如图 4f所示, 在特征值≤0的区域出现2个峰值, 特征值-0.009 a.u表明胞嘧啶取代基—NH2与菲苯核π电子之间的静电引力作用;而更强的引力特征值 (-0.016 a.u) 则证实了嘧啶CN6环富电子域与菲弧角位碳原子与间发生“赠电子-吸电子”引力作用.另外, 虽然胸腺嘧啶比胞嘧啶多一个羰基取代基, 但两者与菲的作用展示了类似的结果.如图 4g所示, 胸腺嘧啶对菲的宏观吸附可与胸腺嘧啶CN6环-菲弧角位碳原子 (粉色箭头所示) 间的“赠电子-吸电子”静电引力相联系.特征值-0.016 a.u位置表明了该静电引力作用 (图 4h).而相对较弱的引力 (-0.009 a.u) 则与胸腺嘧啶环上N取代基与菲π电子间的静电引力有关.
3.3 酸碱对非共价结合的影响图 5为酸碱环境对基因碱基-菲相互作用的影响.表 1则列出了图 2和图 5所显示的pH=5.0、7.0和9.0影响下的吸附参数.对于腺嘌呤-菲相互作用, pH=5.0和9.0下线性指数分别是0.95和1.36, 而中性环境下 (pH=7.0) 为1.64.显然, 酸性环境下, 腺嘌呤对菲的吸附属于分配作用 (1/n=1.0);在中性和碱性条件下, 更高的1/n值则表明腺嘌呤和菲之间的引力变弱 (近藤精一, 2006).因此, 相较于中性和碱性条件, 酸性条件有利于腺嘌呤与菲的结合.
 |
| 图 5 酸碱性对基因碱基-菲相互作用的影响 (a.腺嘌呤,b.鸟嘌呤, c.胞嘧啶,d.胸腺嘧啶) Fig. 5 Effect of acidity and alkalinity on base-phenanthrene interaction |
| 表 1 芳烃菲在4种基因碱基上的Freundlich吸附参数 Table 1 Parameters of the Freundlich equation for aromatic phenanthrene adsorption in four genetic bases |
图 5b为酸碱影响下鸟嘌呤对菲的吸附结果.酸性条件下 (pH=5.0) 的吸附斜率高于碱性环境 (pH=9.0).表 1表明在酸性 (pH=5.0)、中性 (pH=7.0)和碱性 (pH=9.0) 条件下, 鸟嘌呤对菲吸附的线性指数分别为0.96、0.92和0.68.显然, 在酸性和中性环境下, 线性指数 (1/n) 接近于1.0, 为分配吸附.而在碱性环境下线性指数为0.68, 远小于1.0.更低的线性指数值表明两分子间存在促进吸附的引力作用 (近藤精一, 2006).显然, 碱性条件下鸟嘌呤-菲间亲和力更强.这可能与碱性环境下鸟嘌呤氧取代基的脱质子化过程有关.根据路易斯酸碱理论 (Pearson, 1963), 高pH值使作为电子赠体的鸟嘌呤脱质子化, 表面负电性增强, 则整个分子极化减弱, 母体结构上的电子更易于与菲发生π-π引力作用.
图 5c为酸碱性影响下胞嘧啶对菲的等温吸附曲线, 表 1展示了对应拟合后相关参数.在pH=5.0、7.0和9.0的条件下, 胞嘧啶对菲吸附的线性指数分别是1.52、0.96和1.10.在中性和碱性环境下, 线性指数1/n约等于1.0, 为分配吸附, 表明分子间亲和力较强.而在酸性环境下 (pH=5.0), 1/n的高值 (1.52) 则表明胞嘧啶-菲间的引力变弱 (近藤精一等, 2006);显然, 中性和碱性环境更有利于胞嘧啶与菲结合.同时, 酸性、中性和碱性条件下, 胸腺嘧啶对菲吸附的线性指数分别为1.40、1.16和1.07.中性和碱性环境下, 吸附类型为分配作用;酸性的条件不利于胸腺嘧啶对菲的吸附.同样, 该结论可利用“路易斯酸碱-极化”的理论进行解释, 中性或碱性条件下, 作为电子给予体含氧嘧啶脱质子化, 表面负电性增强, 则整个分子极化减弱, 平面π电子更易于与菲弧角位碳原子发生“赠电子-吸电子”作用, 分子亲和力增强.
3.4 结合能依赖性对计算与实验获得的结合能的结果进一步分析, 目的是证明模型计算对碱基作用位点预测的可靠性.针对每一个吸附样点, 本文计算了分配系数Kd:
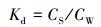
|
(3) |
对于每一组吸附数据, 根据设置样点数, 计算出6个分配系数;这6个分配系数 (Kd) 界定了分配数据的可允许误差范围.基于分配系数, 计算结合能量 (ΔG0):
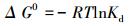
|
(4) |
式中, R取值为8.314 J·mol-1·K-1;T是热力学温度, 取值为298.15 K.
表 2为计算的碱基对菲的分配常数Kd和对应结合能.菲-腺嘌呤分配系数范围为51.21~135.94 L·mol-1, 对应结合能为-9.76~-12.18 kJ·mol-1;菲-鸟嘌呤分配系数范围为87.15~102.77 L·mol-1, 对应的结合能范围为-11.07~-11.48 kJ·mol-1;菲-胞嘧啶分配系数范围为56.88~66.97 L·mol-1, 结合能范围为-10.02~-10.42 kJ·mol-1;菲-胸腺嘧啶分配系数范围为20.45~27.33 L·mol-1, 结合能范围为-7.48~-8.20 kJ·mol-1.值得注意的是, 结合能的负号表示该反应是自发的反应过程.并且所有的结合能低于20 kJ·mol-1, 表明所有碱基对菲的吸附为自发的非共价作用下的物理结合过程 (Gu et al., 1994).
| 表 2 芳烃菲在4种基因碱基上的分配参数 Table 2 Parameters of the aromatic phenanthrene to genetic bases in aqueous solution |
模型计算所获得的结合能 (ΔG0) 公式如下:
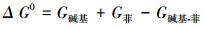
|
(5) |
式中, G碱基、G菲和G碱基-菲分别代表碱基分子、菲分子和吸附完成后碱基-菲的吉布斯自由能.基于公式 (4), 计算获得结合能 (ΔG0) 能换算成分配系数lnKd:
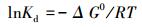
|
(6) |
基于公式 (4)和(6), 把实验获得的分配系数换算成结合能, 同时把计算获得的自由能换算成分配系数, 可以比对计算与实验结果间的吻合度, 结果如图 6所示, 可以看出菲对腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧啶的分配系数 (lnKd) 及对应的结合能.腺嘌呤和鸟嘌呤位于右下角, 与菲的结合能在11 kJ·mol-1以上.胞嘧啶与菲结合能为-10.02~-10.42 kJ·mol-1, 分布于坐标矩阵中心区域;胸腺嘧啶与菲的结合能分布于左上角, 显示了最低的结合能.更重要的是, 通过计算化学所获得的结合能 (黑色星号) 分别对应分布于4种碱基-菲区间, 表明理论计算值与实验值高度吻合, 证实计算模型上基因碱基作用位点的预测的可靠性.
 |
| 图 6 菲与碱基的结合能 Fig. 6 Binding energy between phenanthrene and genetic bases |
1) 在pH=7.0条件下, 腺嘌呤与菲间的引力较弱, 而鸟嘌呤、胞嘧啶和胸腺嘧啶对菲的线性系数约等于1.0, 属于分配作用, 亲和力较高.
2) 酸性条件有利于腺嘌呤对菲的结合 (分配, 1/n=1), 但不利于胸腺嘧啶与菲的结合.中性或碱性条件下, 由于胞嘧啶和胸腺嘧啶存在羰基取代基, 作为电子给予体含氧嘧啶脱质子化, 表面负电性增强, 则整个分子极化减弱, 平面π电子更易于与菲弧角位碳原子发生“电子受体-赠体”间作用, 亲和作用增强.
3) 红外光谱和弱力等值面分析表明, 鸟嘌呤和腺嘌呤通过2组CN环 (CN6环和5环) 同时与菲的2个毗邻苯核发生π-π相互作用;而2类嘧啶仅通过分子平面与菲弧角位置的碳原子发生“赠电子-吸电子”静电引力作用.能量计算的结果表明, 基因碱基作用位点的预测结果是可信的, 属于自发的非共价键成因的物理结合过程.该研究可为深入揭示芳香烃基因污染原理提供理论基础, 对于防治芳香烃污染、保障生态安全有重要意义.
| [${referVo.labelOrder}] | Allouche A. 2011. Gabedit-a graphical user interface for computational chemistry softwares[J]. Journal of Computational Chemistry, 32(1) : 174–182. DOI:10.1002/jcc.v32:1 |
| [${referVo.labelOrder}] | Andrushchenko V, Leonenko Z, Cramb D, et al. 2002. Vibrational CD (VCD) and atomic force microscopy (AFM) study of DNA interaction with Cr3+ ions:VCD and AFM evidence of DNA condensation[J]. Biopolymers, 61(4) : 243–260. DOI:10.1002/(ISSN)1097-0282 |
| [${referVo.labelOrder}] | Chen W, Duan L, Zhu D Q. 2007. Adsorption of polar and nonpolar organic chemicals to carbon nanotubes[J]. Environmental Science & Technology, 41(24) : 8295–8300. |
| [${referVo.labelOrder}] | De Voe H, Wasik S P. 1984. Aqueous solubilities and enthalpies of solution of adenine and guanine[J]. Journal of Solution Chemistry, 13(1) : 51–60. DOI:10.1007/BF00648591 |
| [${referVo.labelOrder}] | Frisch M J T, Schlegel G W, Scuseria H B, et al.2009.Gussian 09, Revision E.01[CD].Wallingford CT:Gaussian, Inc. |
| [${referVo.labelOrder}] | Gu B, Schmitt J, Chen Z, et al. 1994. Adsorption and desorption of natural organic matter on iron oxide:mechanisms and models[J]. Environmental Science & Technology, 28(1) : 38–46. |
| [${referVo.labelOrder}] | Hu X J, Kang F X, Gao Y Z, et al. 2015. Bacterial diversity losses:A potential extracellular driving mechanism involving the molecular ecological function of hydrophobic polycyclic aromatic hydrocarbons[J]. Biotechnology Reports, 5 : 27–30. DOI:10.1016/j.btre.2014.11.002 |
| [${referVo.labelOrder}] | Hu X, Liu J, Mayer P, et al. 2008. Impacts of some environmentally relevant parameters on the sorption of polycyclic aromatic hydrocarbons to aqueous suspensions of fullerene[J]. Environmental Toxicology and Chemistry, 27(9) : 1868–1874. DOI:10.1897/08-009.1 |
| [${referVo.labelOrder}] | Johnson E R, Keinan S, Mori Sanchez P, et al. 2010. Revealing noncovalent interactions[J]. Journal of the American Chemical Society, 132(18) : 6498–6506. DOI:10.1021/ja100936w |
| [${referVo.labelOrder}] | 近藤精一, 石川达雄, 安部郁夫. 2006. 吸附科学[M]. 北京: 化学工业出版社. |
| [${referVo.labelOrder}] | Kang F X, Ge Y Y, Hu X J, et al. 2016. Understanding the sorption mechanisms of aflatoxin B1 to kaolinite, illite, and smectite clays via a comparative computational study[J]. Journal of Hazardous Materials, 320 : 80–87. DOI:10.1016/j.jhazmat.2016.08.006 |
| [${referVo.labelOrder}] | Kang F X, Hu X J, Liu J, et al. 2015. Noncovalent binding of polycyclic aromatic hydrocarbons with genetic bases reducing the in vitro lateral transfer of antibiotic resistant genes[J]. Environmental Science & Technology, 49(17) : 10340–10348. |
| [${referVo.labelOrder}] | Kang F X, Wang H, Gao Y Z, et al. 2013. Ca2+ promoted the low transformation efficiency of plasmid DNA exposed to PAH contaminants[J]. PloS One, 8(3) : e58238. DOI:10.1371/journal.pone.0058238 |
| [${referVo.labelOrder}] | 李伟芳, 彭跃, 赵丽娟, 等. 2013. 东北地区城市大气颗粒物中多环芳烃的污染特征[J]. 中国环境监测, 2013, 29(1) : 13–17. |
| [${referVo.labelOrder}] | 刘敏, 侯立军, 邹惠仙, 等. 2001. 长江口潮滩表层沉积物中多环芳烃分布特征[J]. 中国环境科学, 2001, 21(4) : 343–346. |
| [${referVo.labelOrder}] | Loprete D, Hartman K. 1993. Conditions for the stability of the B, C, and Z structural forms of poly (dG-dC) in the presence of lithium, potassium, magnesium, calcium, and zinc cations[J]. Biochemistry, 32(15) : 4077–4082. DOI:10.1021/bi00066a032 |
| [${referVo.labelOrder}] | Lu T, Chen F. 2012. Multiwfn:a multifunctional wavefunction analyzer[J]. Journal of Computational Chemistry, 33(5) : 580–592. DOI:10.1002/jcc.v33.5 |
| [${referVo.labelOrder}] | Murray J S, Politzer P. 2011. The electrostatic potential:an overview[J]. Wiley Interdisciplinary Reviews:Computational Molecular Science, 1(2) : 153–163. DOI:10.1002/wcms.19 |
| [${referVo.labelOrder}] | Ouameur A A, Tajmir-Riahi H A. 2004. Structural analysis of DNA interactions with biogenic polyamines and cobalt (Ⅲ) hexamine studied by Fourier transform infrared and capillary electrophoresis[J]. Journal of Biological Chemistry, 279(40) : 42041–42054. DOI:10.1074/jbc.M406053200 |
| [${referVo.labelOrder}] | Pearson R G. 1963. Hard and soft acids and bases[J]. Journal of the American Chemical Society, 85(22) : 3533–3539. DOI:10.1021/ja00905a001 |
| [${referVo.labelOrder}] | Roy Dennington T K, Millam J.2009.GaussView[CD].Shawnee Mission, KS:Semichem Inc. |
| [${referVo.labelOrder}] | Scrocco E, Tomasi J. 1973. The Electrostatic Molecular Potential as a Tool for the Interpretation of Molecular Properties, in New concepts Ⅱ[M]. Berlin: Springer Co.Ltd. |
| [${referVo.labelOrder}] | Stewart J J. 2012. Stewart Computational Chemistry[M]. Colorado: Springs Co.Ltd. |
| [${referVo.labelOrder}] | Taillandier E, Liquier J. 1992. Infrared spectroscopy of DNA[J]. Methods in Enzymology, 211 : 307–335. DOI:10.1016/0076-6879(92)11018-E |
| [${referVo.labelOrder}] | Trott O, Olson A J. 2010. AutoDock Vina:improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading[J]. Journal of Computational Chemistry, 31(2) : 455–461. |
| [${referVo.labelOrder}] | 虞宝珠, 郭海, 吴瑾光. 1986. 聚腺嘌呤核苷酸 (PolyA) 水化过程的傅里叶变换红外光谱研究-Ⅰ[J]. 静态水化过程的特性[J].物理化学学报, 1986, 2(3) : 207–213. |
| [${referVo.labelOrder}] | Zhang Y X, Tao S, Shen H Z, et al. 2009. Inhalation exposure to ambient polycyclic aromatic hydrocarbons and lung cancer risk of Chinese population[J]. Proceedings of the National Academy of Sciences, 106(50) : 21063–21067. DOI:10.1073/pnas.0905756106 |
| [${referVo.labelOrder}] | 赵其国, 骆永明, 滕应. 2009. 中国土壤保护宏观战略思考[J]. 土壤学报, 2009, 46(6) : 1140–1145. DOI:10.11766/trxb200907180315 |
| [${referVo.labelOrder}] | Zhu D Q, Pignatello J J. 2005. Characterization of aromatic compound sorptive interactions with black carbon (charcoal) assisted by graphite as a model[J]. Environmental Science & Technology, 39(7) : 2033–2041. |