 2017, Vol. 37
2017, Vol. 37



2. 华南理工大学教育部工业聚集区域污染控制与修复重点实验室, 广州 510006;
3. 华南理工大学制浆造纸国家重点实验室, 广州 510006
2. The Key Lab of Pollution Control and Ecosystem Restoration in Industry Clusters, Ministry of Education, South China University of Technology, Guangzhou 510006;
3. State Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou 510006
近年来,采用活化过硫酸盐技术降解水环境中的有机污染物已成为科研热点 (Oh et al., 2009; Liang et al., 2008; Wu et al., 2015).过硫酸盐分为过一硫酸盐 (如KHSO4, PMS) 和过二硫酸盐 (如Na2S2O8, PS),通常所说的过硫酸盐是指过二硫酸盐PS.过硫酸盐经过活化作用可以产生具有强氧化能力的硫酸根自由基 (SO4-·,E0为+2.6V)(House, 1962),因而能够将大量的有机污染物氧化为CO2、H2O和无机物或转换为低毒、易降解的小分子物质.其活化作用是指在光、热或过渡金属离子 (Ag+、Fe2+、Fe3+、Mn2+、Cu2+等) 等条件的激发下产生自由基的过程.目前,大部分的研究者主要关注于过硫酸盐被活化的问题,即探索良好的催化剂活化过硫酸盐产生自由基.其中,基于金属材料和金属复合材料作为非均相催化剂的研究甚为广泛 (Shukla et al., 2010; Sun et al., 2015a; Pu et al., 2015; Matzek et al., 2016; Fang et al., 2013),究其原因是因为该类非均相催化剂具有成本较低、催化性能强且重复使用率高等特点.
金属有机骨架 (Metal-Organic Frameworks, MOFs) 是由有机配体和金属离子通过自组装合成的一类新型有机-无机杂化多孔材料 (Kuppler et al., 2009).由于MOFs材料兼具无机物与有机物的特点,具体表现为较高的比表面积、可控的孔道结构及部分不饱和的金属位点和有机位点等,因而适用于多种领域的应用 (Czala et al., 2009; Jiang et al., 2011; Hong et al., 2009),尤其是MOFs具有不饱和金属位点的特点使其在催化领域的应用上占据优势 (Corma et al., 2010).然而,MOFs中的金属离子往往与有机配体或一些客体分子 (如H2O) 充分配位,导致金属离子趋向饱和状态,因而限制了其自身的催化能力 (Alaerts et al., 2006).目前,随着水环境污染问题的加剧,已有大量的研究将MOFs成功应用于过氧化氢高级氧化技术的水处理中 (Liang et al., 2015; Ai et al., 2014; Huang et al., 2015; 饶秋林, 2015).然而,据我们了解,关于MOFs活化过硫酸盐高级氧化技术的研究很少,也就仅有几篇报道 (Lin et al., 2015; Li et al., 2016).另外,相比单一金属有机骨架,对基于双金属离子的MOFs的研究也越来越广泛,究其原因是因为双金属有机骨架具有更强的某些方面应用能力 (Nayak et al., 2011; Burrows et al., 2011).通常合成双金属MOFs的方法有2种,分别为合成前金属掺杂与合成后金属修饰 (Sun et al., 2015b; Zhou et al., 2016).
基于此,本实验首次以铁基金属有机骨架MIL-101(Fe) 为主体,采用合成前金属掺杂的方法,通过引入适量的铜离子来制备铁铜双金属有机骨架,即MIL-101(Fe, Cu),并研究其活化过硫酸盐降解有机污染物的机理.
2 材料与方法 (Materials and methods) 2.1 主要试剂与仪器FeCl3·6H2O、Cu (NO3)2·3H2O、N, N-二甲基甲酰胺 (DMF)、对苯二甲酸 (BDC)、N2S2O8(PS)、罗丹明B (RhB)、无水乙醇、盐酸、冰乙酸、邻菲罗啉、邻苯二甲酸二丁酯 (DBP) 及其他各种试剂均为分析纯.实验过程所有药品的配制均使用超纯水.pH计、高速离心机、紫外分光光度仪、电感耦合等离子体原子发射光谱仪 (ICP-AES) 仪和高效液相色谱仪 (HPLC).
2.2 催化剂的制备MIL-101(Fe) 和MIL-101(Fe, Cu) 的制备方法参考已报道的文献 (Huang et al., 2015; Skobelev et al., 2013),并经过适当的改进.首先,称取一定量的BDC至50 mL的玻璃烧杯中,加入20 mL DMF试剂,并用磁力转子搅动30 min,使BDC充分溶解,浓度达到1.25 mmol·L-1.然后,称取一定量的氯化铁和硝酸铜固体溶解于5 mL DMF试剂中,其中,Fe3+和Cu2+的总量为2.5 mmol·L-1.将含金属离子混合物的DMF溶液逐滴加入上述50 mL玻璃烧杯中,继续磁力搅拌3 h,使各物质混合均匀.之后,将上述体系中的混合液转移至内衬100 mL的聚四氟乙烯高压釜反应器中,将反应器放置在烘箱内反应,温度调节至110 ℃,时间设定为24 h.待反应釜冷却至室温后,产生的黄色固体用高速离心机分离获得.为了去除黄色固体中残留的BDC和DMF,在电动搅拌器的搅动下,用60 ℃的无水乙醇先后2次清洗黄色固体,之后再用去离子水清洗1 h.为了进一步纯化固体,将其置于200 ℃的管式炉中,在氮气气流的保护下烘干4 h,得到的黄色固体即为催化剂.
按上述方法在不同物质的量比Cu/(Fe+Cu) 的配比下合成一系列催化剂, Cu/(Fe+Cu) 分别为0、0.05、0.15、0.25和0.35,各自分别记为MIL-101(Fe)、MIL-101(Fe, Cu)-1#、MIL-101(Fe, Cu)-2#、MIL-101(Fe, Cu)-3#和MIL-101(Fe, Cu)-4#.
2.3 催化剂的表征采用X射线衍射仪 (XRD,D8-ADVANCE,德国Bruker公司) 对催化剂进行物质的相分析;采用傅里叶变换红外光谱仪 (FTIR,Tensor27型,德国Bruker公司) 对催化剂进行官能团分析;采用扫描电子显微镜和其自带的X射线能谱仪 (SEM,德式LEO 1530 VP型和日式S4800) 对催化剂进行表面形貌特征和元素分析.
2.4 催化剂的催化实验量取100 mL 0.2 mmol·L-1的罗丹明B (RhB) 溶液于玻璃锥形瓶中,同时加入一定量的固体催化剂和过硫酸盐,快速置入摇床中,开启催化剂催化降解实验 (转速为150 r·min-1,温度为25 ℃).每隔一定时间取出1 mL水样,再加入4 mL的乙醇-水溶液 (1:3, V/V) 进行淬灭并摇匀.之后,用0.45 μm的滤膜过滤样品得到滤液,用紫外分光光度仪在波长为554 nm下测定滤液中RhB的含量.
初步尝试用催化剂活化过硫酸盐降解邻苯二甲酸二丁酯 (DBP),DBP初始浓度为0.018 mmol·L-1,催化实验步骤同上.采用高效液相色谱仪 (Agilent 1100, USA) 测定溶液中的DBP,其自带反相C18专用柱 (50 mm×4.6 mm, i.d., 5 μm particle size) 对目标物进行分离.流动相为乙腈-水溶液 (90:10,V/V),流速设为0.6 mL·min-1,紫外检测器波长设置228 nm.检出限最低为30 μg·L-1.
2.5 催化剂的稳定性实验为了研究催化剂的稳定性,采用ICP-AES仪对催化剂中的Cu溶出量进行检测,采用邻菲罗啉分光光度法对催化剂中的Fe溶出量进行检测,并进行了催化剂的循环使用试验.
催化剂循环使用实验采用以下方案进行:经过一次完整的催化降解实验后,用0.45 μm的滤膜过滤溶液,滤膜上的固体用无水乙醇多次洗刷,洗刷液再重复过0.45 μm的滤膜; 待洗至滤液无颜色时,用去离子水反复清洗滤膜上的固体; 然后将催化剂于65 ℃的烘箱内干燥4 h以去除乙醇, 干燥后的催化剂重复第一次催化降解实验.
3 结果与讨论 (Results and discussion) 3.1 催化剂的表征图 1为MIL-101(Fe, Cu) 和MIL-101(Fe) 的XRD谱图.MIL-101(Fe) 的谱图峰出现在2θ=8.6°、8.9°、10.2°、10.6°和16.4°附近,与文献 (Skobelev et al., 2013) 所报道的基本相符.相比MIL-101(Fe),MIL-101(Fe, Cu) 的图谱峰位置并没有改变,只是峰强度明显增强.这可能有2种原因:一是少量的Cu2+已成为MIL-101(Fe, Cu) 的金属节点,因为Cu2+与BDC合成的物质在2θ=8°附近具有强而尖锐的XRD峰 (Huang et al., 2015);二是Cu2+也能够与BDC中的COO—进行配位,Fe3+与Cu2+的相互竞争导致峰强度变化 (Ebrahim et al., 2013).
 |
| 图 1 MIL-101(Fe) 与MIL-101(Fe, Cu) 的XRD图谱 Fig. 1 XRD patterns of MIL-101(Fe) and MIL-101(Fe, Cu) |
图 2是催化剂的红外图谱.由图 2可知,MIL-101(Fe) 与MIL-101(Fe, Cu) 的特征峰很相似,表现在1704、1596、1391、1015、747和547 cm-1处,与文献 (Tang et al., 2015) 报道基本相符.但在1596 cm-1和1391 cm-1处也有轻微的差异.其中,1596和1391 cm-1处分别属于BDC中COO—的非对称振动和对称振动,这可能是因为Cu2+与BDC进行配位导致的差异.
 |
| 图 2 MIL-101(Fe) 与MIL-101(Fe, Cu) 的红外图谱 Fig. 2 FTIR patterns of MIL-101(Fe) and MIL-101(Fe, Cu) |
图 3是催化剂的扫描电镜图,MIL-101(Fe, Cu) 与MIL-101(Fe) 在形貌上差异性很大.从图 3a可以看出,MIL-101(Fe) 具有明显棱角的正八面体,可经过铜掺杂后,正八面体变得不规则且部分出现了长块状的形貌.除此之外,随着Cu2+掺杂的越多,其形状越来越不规则,如MIL-101(Fe, Cu)-2#(图 3b) 依然具有正八面体特征,而MIL-101(Fe, Cu)-4#(图 3d) 已完全失去了八面体而呈现杂乱的碎片形貌.目前,也有关于金属掺杂导致MOFs形貌出现类似变化的报道 (Ebrahim et al., 2013; Binh et al., 2014).这主要是因为双金属的引入使得Fe3+与BDC中的羧基配位受到干扰,进而影响晶粒的生长.表 1是MIL-101(Fe, Cu)-4#对应的EDS元素分析结果.很显然,催化剂的铜含量比预先设定的铜掺杂量低很多,这可能是因为Fe与BDC具有非常强的配位能力,使得铜与BDC的配位明显处于弱势 (Binh et al., 2014).
 |
| 图 3 MIL-101(Fe) 与不同铜掺杂量MIL-101(Fe, Cu) 的SEM图谱 (a.MIL-101(Fe), b.MIL-101(Fe, Cu)-2#, c, d.MIL-101(Fe, Cu)-4#) Fig. 3 SEM images analysis of catalysts with different doping load of Cu (a.MIL-101(Fe), b.MIL-101(Fe, Cu)-2#, c, d.MIL-101(Fe, Cu)-4#) |
| 表 1 MIL-101(Fe, Cu)-4#的能谱分析 Table 1 The EDS analysis of MIL-101(Fe, Cu)-4# |
为了进一步了解Cu2+对催化剂结构的影响,本实验测定了MIL-101(Fe)、MIL-101(Fe, Cu)-2#和MIL-101(Fe, Cu)-4#的比表面积,结果如图 4所示.MIL-101(Fe)、MIL-101(Fe, Cu)-2#和MIL-101(Fe, Cu)-4#对应的比表面积分别为294、346和363 m2·g-1,说明Cu2+的掺杂导致BET增大.有趣的是,MIL-101(Fe) 的比表面积远低于众多文献的报道 (Tang et al., 2015; Férey et al., 2005)( > 1000 m2·g-1),然而,Li等 (2016a)的文献研究也出现了类似的结果.出现这种现象可能是因为MIL-101(Fe) 对气体 (N2或O2) 具有选择性吸附作用,因为很多MOFs材料都可以选择性吸附气体从而实现气体分离 (Dinca et al., 2005).
 |
| 图 4 MIL-101(Fe) 与MIL-101(Fe, Cu) 的氮气吸附/解析图 Fig. 4 Nitrogen adsorption/desorption isotherms of MIL-101(Fe) and MIL-101(Fe, Cu) |
图 5是催化剂MIL-101(Fe) 与MIL-101(Fe, Cu)-2#活化过硫酸钠降解RhB的结果.首先,MIL-101(Fe) 与MIL-101(Fe, Cu)-2#对RhB的吸附作用分别为16%和25%,很显然,后者的吸附性强于前者.这可能是由于Cu2+的掺杂导致MIL-101的孔径变大且表面积提高,先前的文献也有类似的报道 (Zhou et al., 2016).在RhB/PS体系里,RhB的去除率达到23%,表明PS具有自分解能力而产生SO4-·.在MIL-101(Fe, Cu)-2#/PS/RhB的氧化体系里,当反应120 min时,RhB的去除率就达到95%以上.实验说明合成的催化剂具有较好的催化性能,能够活化过硫酸盐去除有机物.另外,相比MIL-101(Fe),MIL-101(Fe, Cu)-2#的降解速率和降解效果均更有优势,说明铜掺杂具有增强催化能力的作用.
 |
| 图 5 MIL-101(Fe, Cu)-2#与MIL-101(Fe) 对RhB的去除效果 (RhB 0.2 mmol·L-1, 催化剂0.3 g·L-1,PS 8 mmol·L-1, pH不调整) Fig. 5 The removal rate of RhB by MIL-101(Fe, Cu)-2# and MIL-101(Fe) (RhB 0.2 mmol·L-1, catalyst 0.3 g·L-1, PS 8 mmol·L-1, without pH adjustemnt) |
图 6是不同铜掺杂量的催化剂活化过硫酸盐的结果,各催化剂的催化能力为MIL-101(Fe, Cu)-1# < MIL-101(Fe, Cu)-2# < MIL-101(Fe, Cu)-3# < MIL-101(Fe, Cu)-4#;除此之外,各催化剂的吸附能力也有类似的结果.说明铜掺杂量越多,吸附能力越强,催化效果也越好.结合以上材料的扫面电镜图,将其归结于以下几个原因:①铜离子的引进影响了合成过程中催化剂的晶粒生长,表现为形貌特征的差异,铜离子引入越多,差异越大,SEM图也说明了这种现象;由于形貌上的差异,使得催化剂MIL-101(Fe, Cu)-4#的表面占有优势,因而对RhB具有更强的吸附性,使得自由基在催化剂表明上就可以降解更多的RhB;②除了催化剂本身的铁离子,铜离子也作为新的金属活性位点活化过硫酸盐产生自由基;③铜离子与铁离子都能够与BDC配位,它们之间的相互竞争影响了配位方式,使得原本与BDC中的羧基紧紧配位的铁离子出现了不饱和配位倾向.
 |
| 图 6 不同铜掺杂量的MIL-101(Fe, Cu) 对RhB的去除效果 (RhB 0.2 mmol·L-1, 催化剂0.3 g·L-1,PS 8 mmol·L-1, pH不调整) Fig. 6 Influence of Cu2+ doping of catalysts on the removal rate of RhB (RhB 0.2 mmol·L-1, catalyst 0.3 g·L-1, PS 8 mmol·L-1, without pH adjustemnt) |
考虑到过硫酸盐对催化的影响,实验研究了MIL-101(Fe, Cu)-4#分别在PS为2、4、6和8 mmol·L-1条件下的催化能力.如图 7a所示,PS投加量直接影响了RhB的去除率.当PS投加量从2 mmol·L-1增加至6 mmol·L-1时,反应60 min后,去除率由70%提高到90%,降解速率也明显加快.当PS从6 mmol·L-1增加至8 mmol·L-1时,虽然去除率和降解速率进一步提高,但提高的程度并不大.由于在6 mmol·L-1的过硫酸盐浓度下,反应90 min时,MIL-101(Fe, Cu)-4#对RhB的去除率已经为95%,考虑到继续提高PS的浓度会增加成本,因此,实验选择PS的初始浓度为6 mmol·L-1.
 |
| 图 7 不同PS投加量 (a) 和不同初始pH (b) 对催化剂催化性能的影响 (RhB 0.2 mmol·L-1, 催化剂0.3 g·L-1, PS 6 mmol·L-1) Fig. 7 Influence of PS concentration (a) and initial pH (b) on catalytic performance in persulfate activation (RhB 0.2 mmol·L-1, catalyst 0.3 g·L-1, PS 6 mmol·L-1) |
pH在废水高级氧化处理中是一个重要的参数,pH的大小直接影响催化剂的催化能力.在PS为6 mmol·L-1和MIL-101(Fe, Cu) 为0.3 g·L-1的前提下,调节pH分别至2.68、2.78、2.90、3.20、3.40、4.00、5.00、7.00和8.00,其催化效果如图 7b所示.pH在2.68~3.40内,催化剂的催化效果较好,当pH高于4时,效果明显下降.显然,酸性条件下更有利于催化剂的催化作用.
3.2.4 金属离子的溶出和催化剂的循环使用由3.2.3节研究的pH影响可知,酸性条件更有利于实验的进行,故不得不思考这样一个问题:是不是材料本身在酸性水溶液下的不稳定导致金属离子的溶出,其本身就是金属离子的均相催化呢?因此,对金属离子溶出量的研究是必不可少的.首先,用ICP分别对催化剂固体MIL-101(Fe, Cu)-4#和反应液进行铜检测,结果表明,催化剂固体中铜掺杂量不足1%,反应液的铜离子也只有0.061 mg·L-1.因此,对金属离子的溶出可以只考虑铁离子.本实验采用邻菲罗啉测定反应液的铁含量 (反应条件:0.3 g·L-1MIL-101(Fe, Cu)-4#,6 mmol·L-1PS,pH调至2.68) 和酸性水浸出液中的铁含量 (反应条件:0.3 g·L-1MIL-101(Fe, Cu)-4#,pH调至2.68),结果如图 8所示.由图可知,在150 min时,反应液和酸性水浸出液中的铁含量分别为17.9 mg·L-1与8.7 mg·L-1,可见后者铁含量是前者铁含量的1/2.这说明催化剂并不是因为在溶液中本身的不稳定而导致金属离子溶出,可能是由于催化剂中铁活性中心与过硫酸盐的强劲作用,结构出现不稳定或受损才导致铁溶出.同时,为了检验铁离子的均相催化作用,往酸性水浸出液里同时加入0.2 mmol·L-1RhB和6 mmol·L-1PS开启降解实验,反应150 min后,对RhB的去除率只达到了43%.这说明催化剂在活化PS的过程中,非均相反应占据主导作用,但也包括金属离子的均相催化作用.
 |
| 图 8 不同体系下催化剂的铁溶出情况 Fig. 8 Investigation of iron leaching of catalyst in PS activation and in acid solution without PS |
图 9是催化剂MIL-101(Fe, Cu) 的循环使用次数对催化效果的影响.前2次使用的结果表明, 催化剂对RhB的去除率都有90%以上.第3次使用时,催化性能略微下降,而当第4次使用时,去除率只有70%.因此,催化剂的循环使用次数不应多于4次.催化效果的下降一方面是因为催化剂中铁活性位点的流失,另一方面是因为RhB或RhB降解的中间产物遗留在催化剂表面,降低了金属活性位点与溶液中PS的接触能力.
 |
| 图 9 MIL-101(Fe, Cu) 的循环使用对催化能力的影响 (RhB 0.2 mmol·L-1, 催化剂0.3 g·L-1, PS 6 mmol·L-1) Fig. 9 Influence of recycled catalyst on catalytic performance in persulfate activation (RhB 0.2 mmol·L-1, catalyst 0.3 g·L-1, PS 6 mmol·L-1) |
图 10为MIL-101(Fe) 与MIL-101(Fe, Cu)-4#催化过硫酸盐降解DBP的结果.由图可知,相比RhB的去除情况,催化剂对DBP的去除效果明显减弱,去除率低于70%.然而,考虑到DBP本身的难降解性,催化剂的催化应用还是有一定价值的.180 min时,MIL-101(Fe) 与MIL-101(Fe, Cu)-4#对DBP的吸附效率分别为28%与39%,吸附作用能力类似于对RhB的吸附.然而,在过硫酸盐的存在下,MIL-101(Fe) 与MIL-101(Fe, Cu)-4#对DBP的去除效率分别达到了48%与68%.可见,MIL-101(Fe, Cu)-4#对DBP的去除具有更高的价值.
 |
| 图 10 MIL-101(Fe, Cu)-4#与MIL-101(Fe) 对DBP的去除效果 (DBP 0.018 mmol·L-1, 催化剂0.4 g·L-1, PS 3.6 mmol·L-1, pH不调整) Fig. 10 the removal rate of DBP by MIL-101(Fe, Cu)-4# and MIL-101(Fe) (DBP 0.018 mmol·L-1, catalyst 0.4 g·L-1, PS 3.6 mmol·L-1, without pH adjustemnt) |
MIL-101(Fe, Cu) 的金属活性位点包括≡Fe3+和≡Cu2+.根据ICP的测定结果可知,催化剂固体铜含量不足1%(见3.2.3节),因此,≡Fe3+是主要的金属活性位点,而≡Cu2+主要是改变催化剂形貌的作用,如提高比表面积.此外,相比MIL-101(Fe),MIL-101(Fe, Cu) 的催化能力增强,这说明具有活性位点的铁离子增多.由于Cu2+与Fe3+都能够与BDC配位,它们之间的相互竞争会影响Fe3+的配位方式,使得原本与BDC中的羧基进行紧密配位的Fe3+出现了不饱和配位倾向,进而提高了铁活性位点比例.
目前,已有相关的研究指出≡Fe3+能够催化过硫酸盐产生SO4-·降解污染物.例如, Liu等 (2014)指出, 含3价铁离子的矿物氧化物能够缓慢催化PS产生SO4-·.Liang等 (2009)的研究结果表明,Fe3+与EDTA形成的络合物能够有效降解三氯乙烯,其中的原理在于Fe3+被还原为Fe2+后仍然与EDTA络合,Fe2+催化PS产生自由基.Lin等 (2015)首次报道了铁基金属有机骨架MIL-88A的催化特性,利用其丰富的铁离子位点活化PS产生自由基,进而有效地降解RhB污染物.其反应机理在于S2O82-提供一个电子给MIL-88A,使MIL-88A的≡Fe3+还原为≡Fe2+,即为≡Fe2+@MIL-88A,二价化合态的铁离子迅速与S22-O8反应产生SO4-·.Li等 (2016b)也报道了4种铁基金属有机骨架 (MIL-88B (Fe)、MIL-53(Fe)、MIL-100(Fe) 和MIL-101(Fe)) 催化PS的研究.本实验合成的铁铜双金属有机骨架MIL-101(Fe, Cu) 材料,其催化机理与以上介绍的文献类似,可由式 (1)~(3) 表示.首先,MIL-101(Fe, Cu) 上的铁活性位点 (≡Fe3+) 与过硫酸根离子 (S2O82-) 接触,S2O82-提供一个电子给铁活性位点,自身氧化成S2O8-·,而≡Fe3+被还原为≡Fe2+.产生的≡Fe2+作为MIL-101(Fe, Cu) 的新的活性位点,迅速催化S2O82-产生SO4-·,而其本身被氧化成≡Fe3+,恢复成原来状态.SO4-·迅速攻击有机污染物,破坏其分子结构,使其降解为小分子中间产物或CO2和H2O.
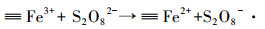
|
(1) |
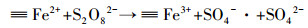
|
(2) |

|
(3) |
1) 采用XRD、FTIR、SEM及EDS等手段对催化剂进行材料表征,结果发现, 相比MIL-101(Fe),Cu2+的掺杂影响了催化剂的晶粒生长,具体表现在XRD特征峰的增强,FTIR特征峰的变化,SEM形貌呈现不规则而杂乱的趋势等.此外,BET结果表明,Cu2+的掺杂有利于提高催化剂比表面积,且随着Cu2+引入量越多,比表面积越大,呈现MIL-101(Fe, Cu)-4# > MIL-101(Fe, Cu)-2# > MIL-101(Fe) 的趋势.
2) 实验结果表明,MIL-101(Fe, Cu)-4#活化PS对有机污染物的去除能力最强.在最佳反应条件下,MIL-101(Fe, Cu)-4#活化PS在90 min时对RhB的去除率为95%,在180 min时对DBP的去除率为68%.
3) pH是影响催化活性的重要因素,MIL-101(Fe, Cu) 在pH为2.68~4.00范围内具有较好的催化能力.通过实验分析,催化剂与PS之间的作用会促进催化剂铁离子的溶出;在MIL-101(Fe, Cu)/PS/RhB的氧化体系中,催化剂可以循环使用4次.
4) 通过比较分析发现,MIL-101(Fe, Cu) 比MIL-101(Fe) 的催化能力明显增强.一方面,Cu2+的引进改变了催化剂的结构,提高了比表面积,进而对污染物具有更好的吸附性,使产生的SO4-·在催化剂表面就可以得到充分利用;另一方面,Cu2+与BDC中羧基的配位,抑制了Fe3+的配位方式,使原本饱和的≡Fe3+趋向于不饱和状态,不饱和的≡Fe3+与PS作用生成≡Fe2+,产生的≡Fe2+催化S2O82-产生SO4-·迅速降解有机污染物.
| [${referVo.labelOrder}] | Ai L H, Zhang C H, Li L L, et al. 2014. Iron terephthalate metal-organic framework:Revealing the effective activation of hydrogen peroxide for the degradation of organic dye under visible light irradiation[J]. Applied Catalysis B:Environmental, 148 : 191–200. |
| [${referVo.labelOrder}] | Alaerts L, Séguin E, Poelman H, et al. 2006. Probing the lewis acidity and catalytic activity of the metal-organic framework[Cu3(btc)2](BTC=Benzene-1, 3, 5-tricarboxylate)[J]. Chemistry-A European Journal, 2(28) : 7353–7363. |
| [${referVo.labelOrder}] | Binh N T, Tien D M, Giang L T K, et al. 2014. Study on preparation and characterization of MOF based lanthanide doped luminescent coordination polymers[J]. Materials Chemistry and Physics, 143(3) : 946–951. DOI:10.1016/j.matchemphys.2013.09.048 |
| [${referVo.labelOrder}] | Burrows A D. 2011. Mixed-component metal-organic frameworks (MC-MOFs):enhancing functionality through solid solution formation and surface modifications[J]. Cryst Eng Comm, 13(11) : 3623–3642. DOI:10.1039/c0ce00568a |
| [${referVo.labelOrder}] | Corma A, Garcia H, Xamena F X L I. 2010. Engineering metal organic frameworks for heterogeneous catalysis[J]. Chemical Reviews, 110(8) : 4606–4655. DOI:10.1021/cr9003924 |
| [${referVo.labelOrder}] | Czala A U, Trukhan N, Müller U. 2009. Industrial applications of metal-organic frameworks[J]. Chemical Society Reviews, 38 : 1284–1293. DOI:10.1039/b804680h |
| [${referVo.labelOrder}] | Dinca M, Long J R. 2005. Strong H2 binding and selective gas adsorption within the microporous coordination solid Mg3(O2C-C10H6-CO2)3[J]. Journal of the American Chemical Society, 127(26) : 9376–9377. DOI:10.1021/ja0523082 |
| [${referVo.labelOrder}] | Ebrahim A M, Bandosz T J. 2013. Ce (Ⅲ) doped Zr-based MOFs as excellent NO2 adsorbents at ambient conditions[J]. ACS Applied Materials & Interfaces, 5(21) : 10565–10573. |
| [${referVo.labelOrder}] | Fang G D, Dionysiou D D, AlAbed S R, et al. 2013. Superoxide radical driving the activation of persulfate by magnetite nanoparticles:Implications for the degradation of PCBs[J]. [J].Applied Catalysis B:Environmental, 129 : 328–332. |
| [${referVo.labelOrder}] | Férey G, Mellot-Draznieks C, Serre C, et al. 2005. A Chromium terephthalate-based solids with unusually large pore volumes and surface area[J]. Science, 309(5743) : 2040–2042. DOI:10.1126/science.1116275 |
| [${referVo.labelOrder}] | Hong D Y, Hwang Y K, Serre C, et al. 2009. Porous chromium terephthalate MIL-101 with coordinatively unsaturated sites:Surfacefunctionalization, encapsulation, sorption and catalysis[J]. Advanced Functional Materials, 19(10) : 1537–1552. DOI:10.1002/adfm.v19:10 |
| [${referVo.labelOrder}] | House D A. 1962. Kinetics and mechanism of oxidations by peroxydisulfate[J]. Chemical Reviews, 62(3) : 185–200. DOI:10.1021/cr60217a001 |
| [${referVo.labelOrder}] | Huang K, X uY, Wang L Q, et al. 2015. Heterogeneous catalytic wet peroxide oxidation of simulated phenol wastewater by copper metal-organic frameworks[J]. RSC Adv, 5(41) : 32795–32803. DOI:10.1039/C5RA01707F |
| [${referVo.labelOrder}] | Jiang H L, Xu Q. 2011. Porous metal-organic frameworks as platforms for functional applications[J]. Chemical Communications, 47 : 3351–3370. DOI:10.1039/c0cc05419d |
| [${referVo.labelOrder}] | Kuppler R J, Timmons D J, Fang Q R, et al. 2009. Potential applications of metal-organic frameworks[J]. Coordination Chemistry Reviews, 253(23/24) : 3042–3066. |
| [${referVo.labelOrder}] | Li H X, Wan J Q, Ma Y W, et al. 2016a. Degradation of refractory dibutyl phthalate by peroxymonosulfate activated with novel catalysts cobalt metal-organic frameworks:Mechanism, performance, and stability[J]. Journal of Hazardous Materials, 318 : 154–163. DOI:10.1016/j.jhazmat.2016.06.058 |
| [${referVo.labelOrder}] | Li X H, Guo W L, Liu Z H, et al. 2016b. Fe-based MOF sfor efficient adsorption and degradation of acid orange 7 in aqueous solution via persulfate activation[J]. Applied Surface Science, 369 : 130–136. DOI:10.1016/j.apsusc.2016.02.037 |
| [${referVo.labelOrder}] | Liang C J, Huang C F, Chen Y J. 2008. Potential for activated persulfate degradation of BTEX contamination[J]. Water ResEarch, 42(15) : 4091–4100. DOI:10.1016/j.watres.2008.06.022 |
| [${referVo.labelOrder}] | Liang C J, Liang C P, Chen C C. 2009. pH dependence of persulfate activation by EDTA/Fe (Ⅲ) for degradation of trichloroethylene[J]. Journal of Contaminant Hydrology, 106(3/4) : 173–182. |
| [${referVo.labelOrder}] | Liang R W, Jing F F, Shen L J, et al. 2015. MIL-53(Fe) as a highly efficient bifunctionalphotocatalyst for the simultaneous reduction of Cr (Ⅵ) and oxidation of dyes[J]. Journal of Hazardous Materials, 287 : 364–372. DOI:10.1016/j.jhazmat.2015.01.048 |
| [${referVo.labelOrder}] | Lin K Y A, Chang H A, Hsu C J. 2015. Iron-based metal organic framework, MIL-88A, as a heterogeneous persulfate catalyst for decolorization of Rhodamine B in water[J]. RSC Advances, 5(41) : 32520–32530. DOI:10.1039/C5RA01447F |
| [${referVo.labelOrder}] | Liu H Z, Bruton T A, Doyle F M, et al. 2014. In situ chemical oxidation of contaminated groundwater by persulfate:decomposition by Fe (Ⅲ)-and Mn (Ⅳ)-containing oxides and aquifer materials[J]. Environmental Science & Technology, 48(17) : 10330–10336. |
| [${referVo.labelOrder}] | Matzek L W, Carter K E. 2016. Activated persulfate for organic chemical degradation:A review[J]. Chemosphere, 151 : 178–188. DOI:10.1016/j.chemosphere.2016.02.055 |
| [${referVo.labelOrder}] | Nayak S, Harms K, Dehnen S, et al. 2011. New three-dimensional metal-organic framework with heterometallic[Fe-Ag] building units:Synthesis, crystalstructure, and functional studies[J]. Inorganic Chemistry, 50(7) : 2714–2716. DOI:10.1021/ic1019636 |
| [${referVo.labelOrder}] | Oh S Y, Kim H W, Park J M, et al. 2009. Oxidation of polyvinyl alcohol by persulfate activated with heat, Fe2+, and zero-valent iron[J]. JHazardMater, 168(1) : 346–351. |
| [${referVo.labelOrder}] | Pu M J, Ma Y W, Wan J Q, et al. 2015. Fe/S doped granular activated carbon as a highly active heterogeneous persulfate catalyst toward the degradation of Orange G and diethyl phthalate[J]. Journal of Colloid and Interface Science, 418 : 330–337. |
| [${referVo.labelOrder}] | 饶秋林. 2015. Fe-MIL-101、Fe-MIL-53及Fe/MIL-125(Ti) 非均相Fenton反应降解罗丹明B溶液[D]. 昆明: 云南大学 |
| [${referVo.labelOrder}] | Shukla P, Wang S B, Singh K, et al. 2010. Cobalt exchanged zeolites for heterogeneous catalytic oxidation of phenol in thepresence of peroxymonosulphate[J]. Applied Catalysis B:Environmental, 99(1/2) : 163–169. |
| [${referVo.labelOrder}] | Skobelev I Y, Sorokin A B, Kovalenko K A, et al. 2013. Solvent-free allylic oxidation of alkenes with O2 mediated by Fe-and Cr-MIL-101[J]. Journal of Catalysis, 298 : 61–69. DOI:10.1016/j.jcat.2012.11.003 |
| [${referVo.labelOrder}] | Sun C, Zhou R, E J N, et al. 2015a. Magnetic CuO@Fe3O4 nanocomposite as a highly active heterogeneous catalyst of persulfate for 2, 4-dichlorophenol degradation in aqueous solution[J]. RSC Advances, 5(70) : 57058–57066. DOI:10.1039/C5RA09821A |
| [${referVo.labelOrder}] | Sun Z G, Zhang Y, Liu H O, et al. 2015b. Ag-Cu-BTC prepared by postsynthetic exchange as effective catalyst for selective oxidation of toluene to benzaldehyde[J]. Catalysis Communications, 59 : 92–96. DOI:10.1016/j.catcom.2014.09.047 |
| [${referVo.labelOrder}] | Tang J, Yang M, Yang M, et al. 2015. Heterogeneous Fe-MIL-101 catalysts for efficient one-pot four-component coupling synthesis of highly substituted pyrroles[J]. New Journal of Chemistry, 39(6) : 4919–4923. DOI:10.1039/C5NJ00632E |
| [${referVo.labelOrder}] | Wu Y L, Bianco A, Brigante M, et al. 2015. Sulfate radical photogeneration using Fe-EDDS:Influence of critical parameters and naturally occurring scavengers[J]. Environmental Science & Technology, 49(24) : 14343–14349. |
| [${referVo.labelOrder}] | Zhou Z Y, Mei L, Ma C, et al. 2016. A novel bimetallic MIL-101(Cr, Mg) with high CO2 adsorption capacity and CO2/N2 selectivity[J]. Chemical Engineering Science, 147 : 109–117. DOI:10.1016/j.ces.2016.03.035 |