 2017, Vol. 37
2017, Vol. 37



化学链燃烧 (Chemical looping combustion, CLC) 作为一种高效近零排放的燃烧技术受到了广泛的关注 (Ishida et al., 1996;Richter et al., 1983;Yang et al., 2008;郑敏等, 2009).其原理是借助于载氧体的作用将传统的燃料与空气直接接触的燃烧反应分解成两个独立的氧化还原反应.如图 1所示, 载氧体 (MxOy) 进入燃料反应器 (FR) 将燃料氧化成的CO2和H2O, 自身被还原 (MxOy-1) 并进入空气反应器 (AR) 氧化再生.在CLC过程中, 燃料不与空气接触, 燃料反应器烟气中只含有CO2和H2O, 无需额外耗能即可获得高浓度的CO2.此外, 还能降低NOx排放, 实现能源的梯级利用, 因此具有广阔的发展前景 (Luo et al., 2014;Moghtaderi et al., 2012;Zheng et al., 2015;Zhou et al., 2015).
 |
| 图 1 化学链燃烧系统概念图 Fig. 1 Schematic progress of chemical looping combustion (CLC) |
在CLC系统中, 载氧体的性能决定了化学链燃烧反应效率和经济性.目前, Cu、Ni、Fe、Mn和Co等金属氧化物被广泛用作载氧体应用于化学链燃烧中 (Cho et al., 2004;黄梅等, 2015).其中CuO、NiO和CoO等虽具有较高反应活性, 但却也各有缺陷:CuO熔点较低, 易烧结 (Hossain et al., 2008;Zhou et al., 2015);NiO热稳性较差, 对环境有污染 (Blas et al., 2015;Hamers et al., 2015);CoO成本较高经济性差 (Mukherjee et al., 2015;覃吴等, 2016).与这几种载氧体相比, Fe2O3具有热稳定性好、载氧高、价格低、环境友好及积碳较少等优点, 从而倍受青睐 (Abad et al., 2007;Tan et al., 2012;Tong et al., 2013;Zhang et al., 2015).但是, Fe2O3反应活性和氧传递效率较低, 严重影响化学链燃烧反应效率 (Cabello et al., 2014;Dong et al., 2012).因此, 国内外学者通过改进制备方法、添加惰性载体和活性成分等来提高铁基载氧体反应活性、载氧能力和热稳定性, 并取得了显著的成果 (Gayán et al., 2012;Zhang et al., 2015).然而, 由于对Fe2O3本身的表面电子结构以及与添加成分的协同作用缺乏详细的机理研究, 限制了铁基载氧体性能的进一步优化.
在催化和燃烧领域, 金属氧化物的微观晶面对其表面反应活性具有巨大影响 (Cai et al., 2014;Hans Joachim et al., 1996;McBriarty et al., 2012;Song et al., 2013;Xiao et al., 2012).Fe2O3(001) 和Fe2O3(1-1 2) 是氧化铁自然裸露的2个主要表面, 许多研究表明, 这2个表面性能具有较大差异.例如, Dzaze等 (2014) 研究了苯在Fe2O3的 (001) 和 (1-1 2) 表面的吸附机理, 指出苯在 (1-1 2) 的吸附构型更加稳定.在化学链燃烧领域, Fe2O3(001) 表面上的CH4、CO、H2S等小分子的吸附、氧化、分解等已有详细报道, 但关于Fe2O3(1-1 2) 表面上的微观吸附-氧化机理研究目前仍较少.本课题组团队前期虽然简单对比了CO与Fe2O3的 (001) 和 (1-1 2) 两个表面的反应性能差异 (Dong et al., 2011), 但Fe2O3(1-1 2) 表面不同配位数原子对CO的吸附、氧化性能产生的影响机理尚不清楚, 阻碍我们获得该表面准确的构效关系, 不利于从表面调控上对载氧体反应性能进行提升.
本文基于密度泛函理论利用CASTEP计算模块, 构建了Fe2O3(1-1 2) 表面模型, 研究表面全部5种不同配位数原子 (记为O2f、O3f、O4f、Fe4f和Fe5f) 的电子结构特性, 对比分析配位数对Fe、O原子电子特性的影响, 并以CO为探针构建5种吸附构型, 研究CO在5种不同配位数原子上生成CO2的反应路径、反应能垒和过渡态.由于铁基载氧体与燃料的反应是以气-固或固-固接触反应为主, 其表面性质对化学反应活性有着关键性的作用 (Hans Joachim et al., 1996;Zeng et al., 2012).因此, 通过研究铁基载氧体的微观结构和表面性质以及燃料在表面的吸附和反应机理, 探索提高其反应活性和载氧能力的方法具有重要的意义.
2 计算方法 (Computational methods)α-Fe2O3属于六方晶系, 每个晶胞呈菱形对称, 体相结构中氧原子形成被扭曲的六方最密堆积原子层, 铁原子则占据其中2/3的八面体空隙, 属R-3c空间群 (Schedel-Niedrig et al., 1995).晶胞参数为:a=b=5.035 Å, c=13.747 Å;α=β=90°, γ=120°(Wang et al., 1998).在Fe2O3斜方六面体晶胞结构中, Fe原子在z轴方向上的自旋态设定为 (+, -, -, +), 其中+和-分别对应自旋向上和自旋向下.以该自旋态设定进行几何优化得到Fe2O3斜方六面体晶胞结构, 能量最低, 结构最稳定 (Sandratskii et al., 1996;Song et al., 2013;Wong et al., 2011).
沿着 (1-1 2) 方向切割Fe2O3的晶胞, 得到了具有周期性结构的Fe2O3(1-1 2) 表面, 包含12个铁原子和19个O原子.为了消除表面中层与层之间的相互作用, 真空层取12 Å.图 2所示为Fe2O3(1-1 2) 完整表面优化后的超胞结构, 表面有不同配位数的O和Fe原子, 分别记为O2f、O3f、O4f、Fe4f和Fe5f(下标代表原子的配位数).
 |
| 图 2 Fe2O3(1-1 2) 表面的几何优化结构以及5种不同配位数的O和Fe原子 Fig. 2 Optimized structure of Fe2O3 (1-1 2) surface and five different coordinate O and Fe atoms |
基于Materials Studio中的CASTEP (Payne et al., 1992) 模块, 采用密度泛函理论和平板模型相结合的方法计算Fe2O3(1-1 2) 结构特性及CO分子与表面的相互作用机理.体系的交换相关能部分采用广义梯度近似 (Generalized gradient approximation, GGA) (Perdew et al., 1992) 和PBE (White et al., 1994) 方法进行处理.采用DFT+U (U=5.0 eV) 的方法, 修正Fe原子3d轨道的强体系相关能 (Zhang et al., 2016).K点取值分别为 (4×4×1), 截断能采用350 eV.优化计算过程中考虑自旋极化作用, 结构中的原子全部放开优化.结构优化以能量、位移和力收敛为判据, 收敛阈分别为2.0 × 10-5 Ha·atom-1、5.0 × 10-3 Å和4.0 × 10-3 Ha·Å-1.结合能Eb定义为吸附前后各物质总能量的变化, 计算公式见式 (1).
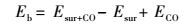
|
(1) |
式中, Esur+CO表示吸附后体系的总能量, Esur和ECO分别表示吸附前表面和自由态时CO分子的能量.
采用线性同步转变 (Linear synchronous transit, LST) 与二次同步转变 (Quadratic synchronous transit, QST) 方法对可能的过渡态进行搜索 (Govind et al., 2003;Zhang et al., 2012).
3 结果与讨论 (Results and discussion) 3.1 Fe2O3(1-1 2) 表面的电子结构特性几何优化后得到Fe2O3(1-1 2) 的纯净表面有5种不同配位数的O和Fe原子位, 如图 2所示.Jiang等 (2011)研究发现表面不同配位数的原子所具有的不同电子特性, 其催化氧化性能也有很大差别.因此, 计算了吸附位原子与相邻原子成键的平均长度, 记为平均键长Lab, 以及吸附位原子的Mulliken (1955)电荷布居.分析可知, O原子位的O—Fe键的平均长度和带负电荷数随着配位数的升高而呈现增长的趋势 (O2f<O3f<O4f), Fe原子位的Fe—O和Fe—Fe键平均长度和带正电荷随着配位数的增加而增加 (Fe4f<Fe5f).固体表面原子键越长与相邻原子间的轨道杂化作用越弱, 其化学稳定性也就越差 (Qin et al., 2015).
根据前线轨道理论, 载氧体与燃料分子的氧化还原反应主要涉及载氧体表面原子的外层电子轨道与燃料分子外层轨道的相互作用 (Chen et al., 2004;Huang et al., 2008).因此, 计算了O原子和Fe原子最外层电子的分波态密度 (PDOS), 如图 3所示.对比O原子的2p轨道可知, O2f、O3f和O4f原子在Fermi能级附近的电子态密度相近, O2f原子在-2~-7 eV之间具有几个尖峰, 说明其局域性较强.Fe4f和Fe5f原子的3d轨道在-7.0~0.0 eV之间都具有较高的离域性, 并且在Fermi能级附近均有较高的电子态密度 (Qin et al., 2012).
 |
| 图 3 Fe2O3(1-1 2) 表面5种不同配位数的O和Fe原子的分波态密度 Fig. 3 PDOS of five different coordinate O and Fe atoms on the pure Fe (1-1 2) surface |
当CO分子以C端垂直接近Fe2O3(1-1 2) 表面原子时, 首先与表面形成物理吸附, 由于只存在相互作用较弱的范德华力, 不能形成稳定的吸附构型.从而将CO进一步接近表面, 相互作用逐渐增强, 经几何优化后发现CO在O2f和O3f位无法形成稳定的化学吸附, 而直接生成了CO2;但在O4f、Fe4f和Fe5f位可以形成稳定的化学吸附.图 4为CO在O2f和O3f位的物理吸附构型以及在O4f、Fe4f和Fe5f位的化学吸附构型.为了分析表面原子的电子特性, 对比分析了吸附CO后各吸附位原子成键的平均长度, 结合能、Mulliken电荷布居, 结果见表 1.
 |
| 图 4 CO在Fe2O3(1-1 2) 表面O和Fe原子位的吸附构型 (a. O2f, b.O3f, c. O4f, d.Fe4f, e. Fe5f) Fig. 4 Adsorption configurations of CO on O and Fe atoms of Fe2O3 (1-1 2) surface (a. O2f, b. O3f, c. O4f, d. Fe4f, e. Fe5f) |
| 表 1 CO吸附前后Fe2O3(1-1 2) 表面吸附位点的键参数、Mulliken电荷布居及CO与表面的距离和结合能 Table 1 Average bond length (Lab) and Mulliken charge population of adsorption sites of the pure surface and adsorbed surface by CO and the distance and bond energy (Eb) between CO and the surface |
图 4a中, CO吸附在表面O2f位点后, 其C—O键 (1.155 Å) 缩短为1.153 Å, 表面O2f原子的平均键长没有变化.图 4b中, CO吸附在表面O3f原子上后, 其C—O键 (1.155 Å) 缩短为1.153 Å, O3f原子的平均键长增加了0.010 Å.图 4c为CO在O4f位的稳定吸附构型, O4f原子形成三键结构, 平均键长增加了0.687 Å, CO分子的C—O被拉长至1.192 Å, 并与O4f原子及相邻的Fe原子成键, 形成二齿形碳酸盐物种.图 4d和4e所示为CO在Fe4f和Fe5f位的吸附构型, CO的C端与Fe4f和Fe5f原子形成C—Fe4f和C—Fe5f键, 键长分别为1.738和1.862 Å.CO的C—O键分别被拉长了0.040 Å和0.020 Å, 说明CO被不同程度的活化.Fe4f和Fe5f位的平均键长分别增加了0.188 Å和0.020 Å.CO在低配位数的O原子吸附时被活化作用较强, 在Fe原子位的活化作用差别不明显.
对比吸附能可知, CO在O2f和O3f位的吸附能远小于CO在O4f、Fe5f和Fe6f位的吸附能.同时, CO在O4f位的结合能是-0.630 eV小于在Fe4f位的结合能 (-0.947 eV) 和在Fe5f位的结合能 (-1.353 eV), 表明CO在Fe2O3(1-1 2) 表面的Fe原子位更容易形成稳定的吸附结构.
以上分析可知, CO与低配位数的O原子和高配位数Fe原子上的相互作用较强 (Cai et al., 2014).Mulliken电荷布居分析显示, CO在O2f和O3f位吸附时, CO的C和O原子电荷从吸附前0.420和-0.420|e|分别变为0.410和-0.400|e|, 0.410和-0.400|e|, 同时O2f和O3f原子所带负电荷均减少了-0.010|e|.在O4f位吸附时, O4f原子所带负电荷分减少了-0.200|e|, CO所带正电荷增加了0.080|e|, CO和O4f原子向表面转移了-0.280|e|.当CO吸附在Fe原子位时, Fe4f和Fe5f所带正电荷均减少了0.070|e|, CO分子中C和O原子所带电荷分别变为0.320和-0.320|e|, 0.290和-0.320|e|, 有少量的电子从CO和Fe吸附位向表面转移.
为了研究吸附反应后CO分子与表面原子的成键机理, 对自由状态下的CO和CO2分子以及CO在5种活性位吸附后的结构进行态密度了分析.自由状态下, CO分子的电子组态为[(1σ)2(2σ)2(3σ)2(4σ)2(1π)4(5σ)2(2π)0] (马淳安等, 2010).图 5a所示为自由CO分子的态密度图, 其中4σ、1π、5σ和2π*轨道分别在-4.95、-2.50、0.00和7.00 eV附近.CO分子的最高占据轨道 (HOMO) 是位于Fermi能级处的5σ轨道, 最低空轨道 (LUMO) 是2π*轨道 (Chen et al., 2004).CO的5σ轨道成分来自于C原子的2s和2p的贡献, 而2π轨道成分则来自于C原子的2p和O原子的2p的贡献.由于5σ和1π轨道之间存在一定的能级差 (2.45eV), 而一般认为1π轨道是定域化的, 不参与成键.
 |
| 图 5 自由CO和CO2以及CO在5种吸附位吸附后的态密度 (a.CO, b. O2f, c.O3f, d.O4f, e.Fe4f, f. Fe5f, g. CO2) Fig. 5 Density states of free CO and CO2 and the adsorbed CO on five adsorption sites (a. CO, b. O2f, c. O3f, d.O4f, e. Fe4f, f. Fe5f, g. CO2) |
图 5b和图 5c为CO在O2f和O3f位反应后的分波态密度, CO分子轨道分别向能量减少的方向移动, 4σ、1π和5σ轨道分别向能量更低处转移, 5σ轨道峰形变宽, 但与自由状态下的CO态密度 (图 5a) 峰形基本相同, 说明与表面之间的存在弱相互作用.图 5d为CO分子吸附在O4f位时态密度, 与自由状态下的CO态密度 (图 5a) 相比较, 在-4~-2.2 eV能量范围内出现了一个明显的峰值, 这是CO2在化学吸附态时表现出的最低非占据轨道 (LUMO) 2πμ的特征峰.并且5σ轨道和1π轨道发生重叠, 重叠部分劈裂出2个新峰, 其中能量在-6.8 eV的峰表现出CO2在化学吸附态时最高占据轨道 (HOMO) 1πg的特征.这2个特征峰的形成表明在此表面产生了碳酸盐物种.
图 5e和5f分别给出了CO分子在Fe4f和Fe5f顶位吸附的分波态密度.吸附作用发生后分子轨道分别向能量减少的方向移动, 4σ、1π、5σ和2π*轨道分别向能量更低处转移, 5σ轨道和1π轨道重叠在-6.5 eV处形成一个新的峰, 这是因为C原子的2π*轨道和Fe原子的3d轨道发生相互杂化的结果.CO的PDOS在吸附发生后均向能量更低处转移, 表明CO分子能稳定吸附在表面.并且自由态下CO分子在费米能级以下以及附近的PDOS的分波态密度峰在吸附以后几乎都消失了, 表明有部分电子从CO分子转移到了表面上, 这个结论和Mulliken原子布居分析相符.CO分子的分波态密度图发生了很大的变化说明了与Fe2O3表面存在强相互作用.
为了更为直观的分析CO与表面间的电荷转移及成键情况, 图 6给出了CO在吸附位的差分电荷密度图.红色代表得到电子, 蓝色代表失去电子.从图中可以看出, CO在O2f和O3f位的吸附后, CO中的C原子呈现蓝色, 相邻O原子呈现红色, 表明C原子中电子向O原子转移, 但与表面O原子几乎不存在电荷的转移.图 6c为CO在O4f位的电荷差分图, 从图中可以看出CO及O4f原子均失去电子, 说明有部分电子向表面转移, 并且C原子和O4f原子之间存在明显的电子云重叠, 它们之间形成的主要是共价键.图 6d和6e所示为CO在Fe4f和Fe5f位吸附时的电荷差分图, 可以看出CO的O原子呈现蓝色, C原子呈现红色, C与Fe原子之间为蓝色, Fe原子呈现红色, 可以推断电子由CO的O原子向C原子转移最终转移到Fe原子上.这是因为CO将HOMO中的电子给予Fe2O3形成键, 同时Fe2O3反馈电子给CO的2π*反键轨道.
 |
| 图 6 CO在O2f(a)、O3f(b)、O4f(c)、Fe4f(d) 和Fe5f(e) 位点化学吸附生成物的电荷差分密度 (红色代表得到电子, 蓝色代表失去电子) Fig. 6 Charge density differences plot relative to CO chemisorbed on O2f (a), O3f (b), O4f (c), Fe4f (d) and Fe5f (e) (The red contours indicate electron density increase and blue contours indicate electron density decrease) |
化学链燃烧过程中CO与铁基载氧体的反应为气-固接触反应, 因此, CO的在Fe2O3表面的氧化反应需经过吸附、氧化及CO2解离3个阶段.CO在表面表面形成稳定吸附后, 与表面O或Fe原子进一步氧化, 最终生成CO2并脱离面.因此, 在计算过渡态时, 以CO在表面的稳定吸附构型为始态 (IS), 以生成CO2的表面为终态 (FS), 利用LST/QST方法对过渡态进行了搜索, 其反应路径和能量变化如图 7所示.
 |
| 图 7 CO在Fe2O3(1-1 2) 表面O2f(a)、O3f(b)、O4f (c)、Fe4f(d) 和Fe5f(e) 位点吸附生成CO2的结构变化、反应路径和能垒 (IS、TS、M和FS分别对应反应初始态、过渡态、中间产物和终态) Fig. 7 Structures of IS, TS, M and FS for CO2 formation by adsorbing CO on O2f (a), O3f (b) and O4f (c), Fe4f (d) and Fe5f (e) sites of the Fe2O3(1-1 2) surface and the corresponding reaction path and potential energy diagram |
图 7a~7c为CO以C端吸附在Fe2O3(1-1 2) 表面O顶位形成CO2分子的反应路径、能量变化和过渡态 (TS).CO分子与表面的O2f和O3f原子难以形成稳定的化学吸附构型, 当CO靠近O2f和O3f原子时, 由于C原子的2p轨道和O的2p轨道间强相互作用, 而直接生成了CO2分子.图 7a中, CO吸附在表面O2f位点后, 其C—O键 (1.155 Å) 被拉长至1.177Å, 表面O2f原子脱离表面与CO分子中的C原子形成C—O2f键 (1.182 Å), 生成物O—C—O2f的键角为179.216°, 其结构非常接近CO2分子.图 7b中, CO吸附在表面O3f原子上后, O3f原子脱离表面与CO分子形成新的化学键C—O3f.其C—O和C—O3f键长分别为1.175 Å和1.185 Å, O—C—O3f的键角为179.285°, 形成化学吸附态的CO2分子结构.
因此, 在做过渡态分析时, 分别以CO分子在O2f和O3f原子顶位上的物理吸附构型作为IS.经过过渡态搜索, 计算结果得到CO在O2f位的反应活化能是3.657 eV, 反应能是-2.868 eV.过渡态中表面O原子与CO成键, 形成CO2化学吸附结构.CO在O3f位点的反应活化能是3.401 eV, 反应能为和-5.186 eV, 过渡态中表面O原子与表面Fe原子成键在CO的作用下被拉长, 与表面Fe原子的三键结构断裂成一键结构.CO在O2f和O3f反应生成CO2的过程中, 没有中间产物生成, 因此反应为一步反应.
CO在表面O4f原子形成化学吸附时, 其C端分别与O4f和Fe成键形成二齿形碳酸盐物种.为了研究此碳酸盐物种的形成过程, 以CO在O4f位的物理吸附构型为始态, 二齿形碳酸盐物种的体系为中间态, 进行了过渡态的搜索.从始态到中间态, CO的C—O键以及Fe—O键均得到活化, 并形成C—O4f和C—Fe键, 此过程需要克服1.864 eV的能垒, 过渡态TS1的结构与中间产物结构类似;二齿形碳酸盐进一步与表面发生反应, O4f—Fe键和C—Fe键断裂形成CO2分子, 此过程需要越过1.097 eV的能垒达到终态, 整个反应过程放热4.474 eV.
图 7e~7f为CO在表面Fe4f和Fe5f原子顶位吸附后生成CO2的反应路径、能量变化和过渡态.CO与Fe原子形成C—Fe键, 形成相对稳定的化学吸附态, 随着反应的进行, CO的C端会与表面Fe原子相邻的O原子结合形成二齿形碳酸盐物种, 此物种从表面解离后生成CO2.因此, 在过渡态分析时, 以CO在表面Fe顶位的稳定吸附构型为始态, 以形成二齿形碳酸盐物种的体系为中间态, 以最终形成CO2分子的体系为终态.CO在表面上的Fe4f和Fe5f原子上分别需要克服0.416 eV和0.219 eV的能垒越过过渡态 (TS1) 生成二齿形碳酸盐物种, 然后此物种分别需要克服0.219 eV和1.462 eV的能垒越过过渡态 (TS2) 形成CO2, 整个反应过程为放热反应分别为-1.106和-2.545 eV.
综上所述, CO在低配位O原子 (O2f和O3f) 位的氧化反应物中间物生成, 形成CO2需要克服的能垒较高.CO在O4f、Fe4f和Fe5f上氧化过程均需经过中间产物, 其中在O4f的活化能比在Fe原子上的活化能高.Huang等 (2015)研究表明, 在Fe2O3(001) 表面CO与表面O原子反应生成CO2的速率是化学链反应的速控步.前文分析的Fe原子在Feimi能级附近具有较好的离域性以及较高的结合能的分析一致.因此, 可以认为高配位数的Fe原子具有更高的氧化性, 在化学链燃烧过程中充当了活性位的作用.
4 结论 (Conclusions)本文利用密度泛函理论研究了Fe2O3(1-1 2) 电子结构特性以及CO在其表面的吸附和氧化反应机理.研究对比分析了表面裸露的所有不同配位数原子的电子特性, 及其吸附-氧化CO的反应机理.结果显示, CO在低配位数的O原子位难以形成稳定的化学吸附, 而是直接由物理吸附生成CO2, 反应遵循一步反应机理, 但是反应活化能较高.而高配位数的O原子位点氧化CO的反应遵循两步反应机理, 即CO在这些位点吸附生成二齿形碳酸盐吸附物, 随后碳酸盐物种脱附生成CO2.结果显示, 高配位数的Fe4f或Fe5f, 由于其较高的氧化态, 在化学链燃烧过程中充当活性位的作用.低配位数的O原子较高的活化能, 是影响铁基载氧体反应活性较低的因素之一, 因此, 下一步的研究将围绕掺杂活性组分来降低低配位O原子的活化能的反应机理.本研究有助于了解铁基载氧体表面化学链燃烧反应的微观动力学过程, 并为载氧体表面结构性能调控制备提供理论借鉴.
| [${referVo.labelOrder}] | Abad A, Mattisson T, Lyngfelt A, et al. 2007. The use of iron oxide as oxygen carrier in a chemical-looping reactor[J]. Fuel, 86(7/8) : 1021–1035. |
| [${referVo.labelOrder}] | Blas L, Dorge S, Michelin L, et al. 2015. Influence of the regeneration conditions on the performances and the microstructure modifications of NiO/NiAl2O4 for chemical looping combustion[J]. Fuel, 153 : 284–293. DOI:10.1016/j.fuel.2015.03.015 |
| [${referVo.labelOrder}] | Cabello A, Abad A, Garcia-Labiano F, et al. 2014. Kinetic determination of a highly reactive impregnated Fe2O3/Al2O3 oxygen carrier for use in gas-fueled Chemical Looping Combustion[J]. Chemical Engineering Journal, 258 : 265–280. DOI:10.1016/j.cej.2014.07.083 |
| [${referVo.labelOrder}] | Cai X, Wang X, Guo X, et al. 2014. Mechanism study of reaction between CO and NiO (001) surface during chemical-looping combustion:Role of oxygen[J]. Chemical Engineering Journal, 244 : 464–472. DOI:10.1016/j.cej.2014.01.079 |
| [${referVo.labelOrder}] | Chen Y, Cao D, Jun Y, et al. 2004. Density functional theory study of CO adsorption on the Fe (111) surface[J]. Chemical Physics Letters, 400(1/3) : 35–41. |
| [${referVo.labelOrder}] | Cho P, Mattisson T, Lyngfelt A. 2004. Comparison of iron-, nickel-, copper-and manganese-based oxygen carriers for chemical-looping combustion[J]. Fuel, 83(9) : 1215–1225. DOI:10.1016/j.fuel.2003.11.013 |
| [${referVo.labelOrder}] | Dong C, Sheng S, Qin W, et al. 2011. Density functional theory study on activity of α-Fe2O3 in chemical-looping combustion system[J]. Applied Surface Science, 257(20) : 8647–8652. DOI:10.1016/j.apsusc.2011.05.042 |
| [${referVo.labelOrder}] | Dong C Q, Liu X L, Qin W, et al. 2012. Deep reduction behavior of iron oxide and its effect on direct CO oxidation[J]. Applied Surface Science, 258(7) : 2562–2569. DOI:10.1016/j.apsusc.2011.10.092 |
| [${referVo.labelOrder}] | Dzade N, Roldan A, de Leeuw N. 2014. A density functional theory study of the adsorption of benzene on hematite (α-Fe2O3) Surfaces[J]. Minerals, 4(1) : 89–115. DOI:10.3390/min4010089 |
| [${referVo.labelOrder}] | Gayán P, Pans M A, Ortiz M, et al. 2012. Testing of a highly reactive impregnated Fe2O3/Al2O3 oxygen carrier for a SR-CLC system in a continuous CLC unit[J]. Fuel Processing Technology, 96 : 37–47. DOI:10.1016/j.fuproc.2011.12.008 |
| [${referVo.labelOrder}] | Govind N, Petersen M, Fitzgerald G, et al. 2003. A generalized synchronous transit method for transition state location[J]. Computational Materials Science, 28(2) : 250–258. DOI:10.1016/S0927-0256(03)00111-3 |
| [${referVo.labelOrder}] | Hamers H P, Gallucci F, Williams G, et al. 2015. Experimental demonstration of CLC and the pressure effect in packed bed reactors using NiO/CaAl2O4 as oxygen carrier[J]. Fuel, 159 : 828–836. DOI:10.1016/j.fuel.2015.07.034 |
| [${referVo.labelOrder}] | Hans-Joachim F, Helmut K, Volker S. 1996. Oxide surfaces[J]. Reports on Progress in Physics, 59(3) : 283. DOI:10.1088/0034-4885/59/3/001 |
| [${referVo.labelOrder}] | Hossain M M, de Lasa H I. 2008. Chemical-looping combustion (CLC) for inherent CO2 separations-a review[J]. Chemical Engineering Science, 63(18) : 4433–4451. DOI:10.1016/j.ces.2008.05.028 |
| [${referVo.labelOrder}] | Huang L, Tang M, Fan M, et al. 2015. Density functional theory study on the reaction between hematite and methane during chemical looping process[J]. Applied Energy, 159 : 132–144. DOI:10.1016/j.apenergy.2015.08.118 |
| [${referVo.labelOrder}] | Huang M, Fabris S. 2008. CO adsorption and oxidation on ceria surfaces from DFT+U calculations[J]. The Journal of Physical Chemistry C, 112(23) : 8643–8648. DOI:10.1021/jp709898r |
| [${referVo.labelOrder}] | 黄梅, 李孔斋, 郑敏, 等. 2015. 煤化学链燃烧钙铁双氧载体竞争还原反应平衡分析[J]. 环境科学学报, 2015, 35(7) : 2030–2036. |
| [${referVo.labelOrder}] | Ishida M, Jin H. 1996. A novel chemical-looping combustor without NOx formation[J]. Industrial & Engineering Chemistry Research, 35(7) : 2469–2472. |
| [${referVo.labelOrder}] | Jiang D E, Dai S. 2011. The role of low-coordinate oxygen on Co3O4(110) in catalytic CO oxidation[J]. Physical Chemistry Chemical Physics:PCCP, 13(3) : 978–984. DOI:10.1039/C0CP01138J |
| [${referVo.labelOrder}] | Luo M, Wang S, Wang L, et al. 2014. Reduction kinetics of iron-based oxygen carriers using methane for chemical-looping combustion[J]. Journal of Power Sources, 270 : 434–440. DOI:10.1016/j.jpowsour.2014.07.100 |
| [${referVo.labelOrder}] | 马淳安, 刘婷, 陈丽涛. 2010. CO和H在Pt/WC (0001) 表面的吸附[J]. 物理化学学报, 2010, 26(1) : 155–162. |
| [${referVo.labelOrder}] | McBriarty M E, Bedzyk M J, Ellis D E. 2012. Structure and properties of a model oxide-supported catalyst under redox conditions:WOx/α-Fe2O3 (0001)[J]. Surface Science, 606(17/18) : 1367–1381. |
| [${referVo.labelOrder}] | Moghtaderi B. 2012. Review of the recent chemical looping process developments for novel energy and fuel applications[J]. Energ Fuel, 26(1) : 15–40. DOI:10.1021/ef201303d |
| [${referVo.labelOrder}] | Mukherjee S, Kumar P, Yang A, et al. 2015. A systematic investigation of the performance of copper-, cobalt-, iron-, manganese-and nickel-based oxygen carriers for chemical looping combustion technology through simulation models[J]. Chemical Engineering Science, 130 : 79–91. DOI:10.1016/j.ces.2015.03.009 |
| [${referVo.labelOrder}] | Mulliken R S. 1955. Electronic Population Analysis on LCAO-MO Molecular Wave Functions[J]. I[J]. The Journal of Chemical Physics, 23(10) : 1833–1840. DOI:10.1063/1.1740588 |
| [${referVo.labelOrder}] | Payne M C, Teter M P, Allan D C, et al. 1992. Iterative minimization techniques for ab initio total-energy calculations:molecular dynamics and conjugate gradients[J]. Reviews of Modern Physics, 64(4) : 1045–1097. DOI:10.1103/RevModPhys.64.1045 |
| [${referVo.labelOrder}] | Perdew J P, Wang Y. 1992. Accurate and simple analytic representation of the electron-gas correlation energy[J]. Physical Review B, 45 : 13244–13249. DOI:10.1103/PhysRevB.45.13244 |
| [${referVo.labelOrder}] | Qin W, Chen Q, Wang Y, et al. 2012. Theoretical study of oxidation-reduction reaction of Fe2O3 supported on MgO during chemical looping combustion[J]. Applied Surface Science, 266 : 350–354. |
| [${referVo.labelOrder}] | Qin W, Lin C F, Long D T, et al. 2015. Reaction activity and deep reduction reaction mechanism of a high index iron oxide surface in chemical looping combustion[J]. Acta Physico-Chimica Sinica, 31(4) : 667–675. |
| [${referVo.labelOrder}] | Richter H J, Knoche K F. 1983. Reversibility of combustion processes, efficiency and costing, second law analysis of processes[C]. ACS Symp. Ser. Washington D.C. Place:Published |
| [${referVo.labelOrder}] | Sandratskii L M, Uhl M, Kübler J. 1996. Band theory for electronic and magnetic properties of α-Fe2O3[J]. Journal of Physics:Condensed Matter, 8(8) : 983. DOI:10.1088/0953-8984/8/8/009 |
| [${referVo.labelOrder}] | Schedel Niedrig T, Weiss W, Schl gl R. 1995. Electronic structure of ultrathin ordered iron oxide films grown onto Pt (111)[J]. Physical Review B, 52(24) : 17449–17460. DOI:10.1103/PhysRevB.52.17449 |
| [${referVo.labelOrder}] | Song J, Niu X, Ling L, et al. 2013. A density functional theory study on the interaction mechanism between H2S and the α-Fe2O3(0001) surface[J]. Fuel Process Technol, 115(0) : 26–33. |
| [${referVo.labelOrder}] | Tan Q, Qin W, Chen Q, et al. 2012. Synergetic effect of ZrO2 on the oxidation-reduction reaction of Fe2O3 during chemical looping combustion[J]. Applied Surface Science, 258(24) : 10022–10027. DOI:10.1016/j.apsusc.2012.06.067 |
| [${referVo.labelOrder}] | 覃吴, 侯翠翠, 张俊姣, 等. 2016. Co-Fe2O3[104]铁基载氧体优化体系作用下褐煤化学链燃烧特性[J]. 化工学报, 2016, 67(4) : 1459–1466. |
| [${referVo.labelOrder}] | Tong A, Bayham S, Kathe M V, et al. 2013. Iron-based syngas chemical looping process and coal-direct chemical looping process development at Ohio State University[J]. Appl Energ(0) : 1836–1845. |
| [${referVo.labelOrder}] | Wang X G, Weiss W, Shaikhutdinov S, et al. 1998. The Hematite (α-Fe2O3) (0001) Surface:Evidence for Domains of Distinct Chemistry[J]. Physical Review Letters, 81(5) : 1038–1041. DOI:10.1103/PhysRevLett.81.1038 |
| [${referVo.labelOrder}] | White J A, Bird D M. 1994. Implementation of gradient-corrected exchange-correlation potentials in Car-Parrinello total-energy calculations[J]. Physical Review B, 50(7) : 4954–4957. DOI:10.1103/PhysRevB.50.4954 |
| [${referVo.labelOrder}] | Wong K, Zeng Q H, Yu A B. 2011. Electronic structure of metal (M=Au, Pt, Pd, or Ru) bilayer modified α-Fe2O3(0001) surfaces[J]. The Journal of Physical Chemistry C, 115(11) : 4656–4663. DOI:10.1021/jp1108043 |
| [${referVo.labelOrder}] | Xiao J, Frauenheim T. 2012. Activation mechanism of carbon monoxide on α-Fe2O3 (0001) surface studied by using first principle calculations[J]. Applied Physics Letters, 101(4) : 041603. DOI:10.1063/1.4739935 |
| [${referVo.labelOrder}] | Yang H, Xu Z, Fan M, et al. 2008. Progress in carbon dioxide separation and capture:A review[J]. Journal of Environmental Sciences, 20(1) : 14–27. DOI:10.1016/S1001-0742(08)60002-9 |
| [${referVo.labelOrder}] | Zeng L, Luo S, Sridhar D, et al. 2012. Chemical looping processes-particle characterization, ionic diffusion-reaction mechanism and reactor engineering[J]. Reviews in Chemical Engineering, 28(1) : 1–42. DOI:10.1515/revce-2011-0010 |
| [${referVo.labelOrder}] | Zhang J J, Qin W, Dong C Q, et al. 2016. Density functional theory study of elemental mercury adsorption on Fe2O3 104 and its effect on carbon deposit during chemical looping combustion[J]. Energ Fuel, 30(4) : 3413–3418. DOI:10.1021/acs.energyfuels.5b01937 |
| [${referVo.labelOrder}] | Zhang X, Song X, Sun Z, et al. 2012. Density functional theory study on the mechanism of calcium sulfate reductive decomposition by carbon monoxide[J]. Industrial & Engineering Chemistry Research, 51(18) : 6563–6570. |
| [${referVo.labelOrder}] | Zhang Y, Doroodchi E, Moghtaderi B. 2015. Comprehensive study of Fe2O3/Al2O3 reduction with ultralow concentration methane under conditions pertinent to chemical looping combustion[J]. Energ Fuel, 29(3) : 1951–1960. DOI:10.1021/acs.energyfuels.5b00080 |
| [${referVo.labelOrder}] | Zhang Y, Doroodchi E, Moghtaderi B. 2015. Reduction kinetics of Fe2O3/Al2O3 by ultralow concentration methane under conditions pertinent to chemical looping combustion[J]. Energ Fuel, 29(1) : 337–345. DOI:10.1021/ef5024252 |
| [${referVo.labelOrder}] | Zheng X, Su Q, Mi W. 2015. Study of a Cu-based oxygen carrier based on a chemical looping combustion process[J]. Energ Fuel, 29(6) : 3933–3943. DOI:10.1021/acs.energyfuels.5b00437 |
| [${referVo.labelOrder}] | Zhou Z, Han L, Nordness O, et al. 2015. Continuous regime of chemical-looping combustion (CLC) and chemical-looping with oxygen uncoupling (CLOU) reactivity of CuO oxygen carriers[J]. Applied Catalysis B:Environmental, 166-167 : 132–144. DOI:10.1016/j.apcatb.2014.10.067 |
| [${referVo.labelOrder}] | 郑敏, 沈来宏, 高正平, 等. 2009. 化学链燃烧中钙基载氧体CaSO4再生的氧化反应机理[J]. 环境科学学报, 2009, 29(2) : 330–338. |