 2017, Vol. 37
2017, Vol. 37



2. 辽河油田特种油开发公司, 盘锦 124000
2. Petrochina Liaohe Oilfield Company, Panjin 124000
随着我国化工行业的快速发展, 天然气化工、石油化工和煤化工规模日益扩大, 化工污水量大幅增加, 因此如何对大量的化工污水进行处理已经成为我国亟需解决的环境问题.苯酚由于其毒性高、有很强的难闻气味(Lin et al., 2003), 并且是化工过程中高分子量的芳香类化合物氧化的中间产物, 因此常被用作高级废水处理的模型化合物(Santos et al., 2001).近些年, 先进氧化技术(AOPs)由于对含有难降解有机物废水的高效处理而受到了人们的关注(王华等, 2007; Yu et al., 2016).在AOPs中, 湿式氧化技术(WAO)已经被广泛用于含酚废水的处理.然而, WAO的反应条件苛刻, 不但能耗高而且对设备材质要求也高, 因此, WAO在实际应用中往往只做预处理技术使用(Debellefontaine et al., 2000; Kim et al., 2011).而利用过渡金属作为催化剂的催化湿式氧化法(CWAO)可以在较低的温度和压力下分解难降解的污染物, 由此降低了反应成本和操作费用(Bhargava et al., 2006; Oliviero et al., 2003).其中由于固体催化剂容易从被处理污水中分离, 而受到了人们的广泛关注.
目前, 用于催化湿式氧化法处理苯酚废水的固体催化剂主要包括:贵金属催化剂, 金属氧化物催化剂和混合氧化物催化剂(Lousteau et al., 2015; Rocha et al., 2015; Yang et al., 2015).Ru、Pd、Pt等贵金属催化剂在催化湿式氧化处理苯酚废水过程中表现出了良好的催化活性.Barbier等(2005)研究表明不同贵金属催化湿式氧化苯酚废水的活性顺序为Ru/CeO2>Pd/CeO2>Pt/CeO2.在160 ℃, 20 bar氧分压的条件下, Ru/CeO2催化剂上苯酚被完全降解.但贵金属催化剂存在价格昂贵, 并且在含卤素、硫、氮化合物的污水中极其容易失活的缺点.因此, 金属氧化物和混合氧化物催化剂被广泛用作贵金属催化剂的替代品(Chang et al., 2005; Chen et al., 2004; 2007).在金属氧化物催化剂和混合氧化物催化剂中, 铜基催化剂表现出较好的氧化活性而被广泛应用于CWAO过程中(Yadav et al., 2014).Kim等(2005)研究了不同过渡金属(Cu、Ni、Co、Fe、Mn)氧化物的催化湿式氧化苯酚废水活性, 结果表明CuO/γ-Al2O3催化剂表现了最佳的催化性能.在铜基催化剂中, 目前以CuO/ZnO/Al2O3催化剂的应用最为普遍, ZnO能够提高铜的分散度和氧化还原能力, Al2O3能够增加表面积防止铜的烧结(Figueiredo et al., 1998).与传统的Al2O3载体相比, MCM-41分子筛、CeO2、CeO2-ZrO2复合氧化物等载体表现出了更好的催化性能(Wu et al., 2001; Keav et al., 2014; Kim et al., 2009).Wu等(2001)研究了介孔材料在催化湿式氧化苯酚废水过程中的作用, 结果表明由于MCM-41分子筛具有均匀的孔道和较大的比表面积而起到了较好的催化效果.Keav等(2014)研究表明CeO2载体的储放氧能力加速了表面物种的吸附速率, 从而改善了催化性能.在CeO2中掺杂Zr4+形成的CeO2-ZrO2复合氧化物固溶体, 可进一步改善CeO2的体相特性, 从而具有更高的储放氧能力和更好的热稳定性(Zhang et al., 2013a; 张磊等, 2012;2015).但CeO2、ZrO2和CeO2-ZrO2载体在苯酚废水氧化过程中所起到的作用还亟需进一步深入研究.因此, 本论文着重探索了CeO2、ZrO2和CeO2-ZrO2载体的结构和特性, 并将其与催化苯酚氧化反应性能相关联, 建立催化剂的构效关系.
2 实验部分(Experimental) 2.1 催化剂的制备不同载体铜基催化剂采用共沉淀法制备(Zhang et al., 2013b; 张磊, 2013).按CuO/ZnO/support(CeO2, ZrO2、CeO2-ZrO2)质量比为45:20:35配制0.1 mol·L-1含Cu、Zn、Ce、Zr的硝酸盐水溶液(中国医药集团上海化学试剂公司, 均为AR)和1.2倍当量0.5 mol·L-1的Na2CO3水溶液(天津市科密欧化学试剂开发中心, AR), 在60 ℃强烈搅拌下, 将碳酸钠滴入硝酸盐混合溶液中, 当pH值达到8时, 停止滴入碳酸钠溶液, 并继续搅拌2 h, 搅拌后在室温下静置12 h.将沉淀抽滤, 用去离子水洗涤, 110 ℃下干燥12 h后在400 ℃下焙烧2 h, 研磨至120目, 压片成型, 粉碎成40~60目的催化剂.CuO/ZnO/CeO2-ZrO2催化剂中, CeO2和ZrO2的物质的量比为1.对比催化剂选用川化股份有限公司催化剂厂生产的质量比为60:25:15的CuO/ZnO/Al2O3工业催化剂.
2.2 催化剂的表征BET比表面积和孔容采用NOVA2200E型(Quantachrome公司)全自动吸附仪测定.催化剂物相分析采用日本理学Ru-200B型X射线粉末衍射仪测定.催化剂的还原性能、铜比表面积(SCu)、铜分散度和储放氧功能(OSC)的测试过程见参考文献(张磊, 2013).X射线光电子能谱(XPS)在VG ESCALAB MK-Ⅱ型(Thermo-VG Scientific公司)光电子能谱仪上进行.采用Al Kα射线激发源(hv=1486.6 eV), 电子结合能(BE)值采用样品的污染碳(C1s =284.6~285.0 eV)作为内标物, 校正样品表面的荷电效应.金属溶出采用Agilent 725 ICP-AES分光光度仪测定.
2.3 催化湿式氧化苯酚实验催化湿式空气氧化苯酚废水实验在带有控温装置的300 mL高压反应釜中进行.将100 mL浓度为100、500、1000和2000 mg·L-1的苯酚溶液和0.5 g催化剂加入到反应釜中, 并控制温度为30 ℃.通入0.8 MPa空气后, 将反应釜加热到一定温度, 以此时的温度和压力作为反应温度和压力, 并作为反应的“零点”计时.此后每隔一定时间取1 mL反应液, 将反应液过滤后采用重铬酸钾法分析化学需氧量(COD)(American Public Health Association (APHA), 2005).COD去除率(X)采用式(1) 计算.
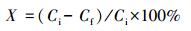
|
(1) |
式中, Ci和Cf分别代表起始和测量样品的COD值.
3 结果与讨论(Results and discussion) 3.1 催化剂物理性质催化剂比表面积、孔容、铜比表面积和铜分散度见表 1.在所制备催化剂中, CuO/ZnO/ZrO2催化剂的比表面积最大, 为143.5 m2·g-1, CuO/ZnO/CeO2催化剂的比表面积最小, 只有50.6 m2·g-1.CuO/ZnO/CeO2-ZrO2催化剂的比表面积(90.8 m2·g-1)与CuO/ZnO/Al2O3工业催化剂的比表面相接近, 略低于CeO2-ZrO2载体的比表面积.催化剂的比表面积直接影响到活性组分的分散情况.与催化剂比表面积相对应, CuO/ZnO/ZrO2催化剂的铜分散度最大, CuO/ZnO/CeO2催化剂的铜分散度最小.虽然CuO/ZnO/Al2O3工业催化剂比表面积略高于CuO/ZnO/CeO2-ZrO2催化剂, 但铜的分散度却低于了CuO/ZnO/CeO2-ZrO2催化的铜分散度, 这是因为两者的组成比例不同所造成的.
| 表 1 催化剂的物理性质 Table 1 Physical characteristics of the catalyst |
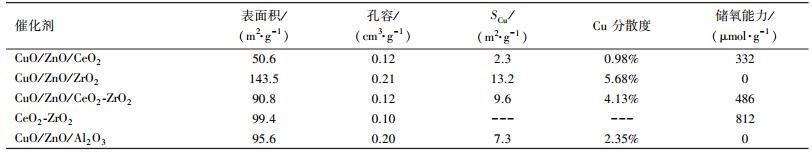
催化剂的储放氧能力(OSC)见表 1.CuO/ZnO/Al2O3工业催化剂和CuO/ZnO/ ZrO2催化剂均没有储放氧能力, 而CuO/ZnO/CeO2催化剂表现出了一定的储放氧能力, 这主要是因为CeO2的储放氧能力产生的.CeO2和ZrO2形成CeO2-ZrO2固溶体后, 能进一步增加储放氧能力, 因此CuO/ZnO/CeO2-ZrO2催化剂的储放氧能力远高于CuO/ZnO/CeO2催化剂.储放氧能力的增加有利于表面氧物种的增多, 而表面氧物种的增多有利于加速苯酚氧化反应过程, 所以增加催化剂储放氧能力有利于苯酚氧化过程的进行.
3.2 催化剂物相组成图 1为不同载体催化剂的XRD谱图.从CuO/ZnO/CeO2催化剂的衍射特征峰可看出, CeO2的衍射特征峰对应的2θ为28.4°.添加ZrO2后, CeO2-ZrO2载体和CuO/ZnO/CeO2-ZrO2催化剂的CeO2和ZrO2的特征峰消失, 并且在2θ=28.9°、33.5°、48.2°和57.1°处出现了明显的CexZr1-xO2固溶体特征衍射峰, 说明样品中CeO2-ZrO2复合氧化物主要以CexZr1-xO2固溶体形式存在.CexZr1-xO2固溶体的形成能提升CeO2的储放氧性能, 从而有利于调节氧化还原反应过程, 提升氧化还原反应速率.除此之外, 除CeO2-ZrO2载体外的样品均存在单斜相的CuO(JCPDS 48-1548) 和六方相的ZnO(JCPDS 36-1451) 衍射特征峰.
 |
| 图 1 催化剂的XRD谱图 Fig. 1 XRD patterns of prepared catalysts |
不同载体催化剂的H2-TPR谱图及用高斯函数进行拟合后的谱图如图 2所示.所制备的催化剂H2-TPR谱图中都出现了一个低温还原峰(肩峰)和一个高温还原峰(主峰), 这是由于不同氧物种的还原过程产生的.还原温度在230~260 ℃之间的肩峰对应表相氧物种的还原, 还原温度在260~300 ℃之间的主峰对应体相氧物种的还原或Cu(Ⅱ)O晶体的还原(Shimokawabe et al., 1990).低温还原峰的出现, 更容易在反应过程中提供表面氧物种, 从而加速氧化反应过程.从图 2中还可以看出, 相比CuO/ZnO/CeO2和CuO/ZnO/ZrO2催化剂, CuO/ZnO/CeO2-ZrO2催化剂的肩峰和主峰都向低温方向移动, 这说明相比CeO2和ZrO2载体, CeO2-ZrO2固溶体载体与活性组分的相互作用更强, 从而使得氧物种更容易被还原, 还原峰温度降低.Monte等(2005)研究表明CeO2-ZrO2固溶体负载过渡金属氧化物催化剂的还原温度越低, 降解苯酚活性越高, 显然的, 这与本实验结果相符.另外, CeO2-ZrO2氧化物固溶体能够活化和转移氧物种.CeO2-ZrO2固溶体在氧化反应过程中能够提供表面氧物种参与反应, 表面氧物种参与反应后, 在CeO2-ZrO2固溶体表面就产生了氧空穴, 氧空穴将反应物氧气活化, 使得氧气转化为活化的表面氧物种继续参与氧化还原反应过程, 这样就加快了氧化还原反应速率.从此说明, CeO2-ZrO2氧化物固溶体是一种良好的氧化还原反应催化剂活性组分或载体.
 |
| 图 2 催化剂的H2-TPR谱图 Fig. 2 H2-TPR profiles of the prepared catalysts |
应用XPS对CuO/ZnO/CeO2-ZrO2催化剂的表面物种及其价态进行了分析.图 3分别为表面Cu 2p的电子结合能谱和表面Ce 3d的电子结合能谱.由图 3a可知, 940~945 eV处出现的Cu 2p振激峰(shake up peak)对应Cu2+的电子结合能, 说明反应前表面铜物种主要以CuO的形式存在, 这与XRD结果相吻合.另外, 由于Ce为镧系过渡金属, 受X光激发后, 产生多电子激发过程, 因此Ce 3d谱峰结构复杂(Mullins et al., 1998).由图 3b可知, 在884.9和903.5 eV处的两个特征峰可归属于Ce3+的电子结合能, 在881.7 eV和916.3 eV处的两个特征峰可归属于Ce4+的电子结合能, 说明Ce3+物种和Ce4+物种共同存于催化剂表面.如前所述, CeO2-ZrO2固溶体能提供表面氧物种和表面氧空穴, 这主要是通过Ce3+和Ce4+的相互转换实现的.表面氧物种的形成有利于苯酚的氧化和分解, 氧空穴的形成有助于反应物O2的解离和产物CO2的形成, 因此, 催化剂表面Ce3+和Ce4+共存有助于协调氧化还原反应过程, 这与H2-TPR结果相符.
 |
| 图 3 CuO/ZnO/CeO2-ZrO2催化剂Cu 2p(a)、Ce 3d(b)和O 1s(c)的XPS谱图 Fig. 3 XPS spectra of Cu 2p(a), Ce 3d (b) and O 1s(c) binding energy region obtained from CuO/ZnO/CeO2-ZrO2 catalyst |
为了进一步说明CuO/ZnO/CeO2-ZrO2催化剂表面氧物种的存在形式, 图 3c为O 1s的电子结合能谱图.从图中可以看出, O 1s存在两个重叠峰, 对应两种不同的氧物种, 低结合能对应的为体相氧物种, 高结合能对应的为表相氧物种(Wang et al., 2008).
3.5 催化湿式空气氧化苯酚性能评价不同反应时间下的催化湿式空气氧化苯酚COD去除率如图 4所示.不加入催化剂的空白实验COD去除率最低, 在反应条件为200 ℃和2 MPa下, 最终(240 min)COD去除率仅为70.6%.CuO/ZnO/Al2O3工业催化剂的最终COD去除率略高于空白实验, 为79.5%.不含CuO的CeO2-ZrO2载体的催化活性高于CuO/ZnO/Al2O3工业催化剂, 说明CeZr载体不仅起到了分散活性组分CuO的作用, 而且也是催化湿式空气氧化苯酚废水的良好催化剂, 这可能是CuO/ZnO/CeO2-ZrO2催化剂具有最佳活性的一个原因.添加CeO2为载体的CuO/ZnO/CeO2催化剂的催化活性进一步提升.尽管CuO/ZnO/CeO2催化剂的Cu分散度最低, 但相比CuO/ZnO/Al2O3工业催化剂, 最终COD值提升了10%.这是因为CeO2具有一定的储放氧能力, 从而提高了催化剂的氧化反应活性.相比Al2O3和CeO2为载体的铜基催化剂, ZrO2为载体的CuO/ZnO/ZrO2催化剂活性更好, 这是因为ZrO2载体不仅改善了Cu的分散, 而且改善了催化剂的还原性能.因此, CuO/ZnO/ZrO2催化剂的最终COD去除率达到了94.9%, 仅略低于CuO/ZnO/CeO2-ZrO2催化剂.从图 4中可以看出, 以CeO2-ZrO2固溶体为载体的CuO/ZnO/CeO2-ZrO2催化剂具有最佳的催化湿式空气氧化苯酚活性, 这是因为以CeO2-ZrO2固溶体为载体不仅能较好的分散CuO物种, 改善催化剂的氧化还原性能, 而且增加了催化剂的储放氧性能, 从而使得CuO/ZnO/CeO2-ZrO2催化剂表现出最佳的催化活性.
 |
| 图 4 催化湿式空气氧化苯酚性能 Fig. 4 CWAO of phenol over the non-catalyst and prepared catalysts |
在CuO/ZnO/CeO2-ZrO2催化剂上, 考察了反应温度及苯酚废水初始浓度对COD去除率的影响, 如图 5所示.由图 5a可知, 在催化湿式空气氧化过程中, 反应温度对COD去除率的影响非常显著.当反应温度为180 ℃时, COD去除率仅为77.5%.当反应温度增加到200 ℃和220 ℃时, COD去除率分别达到了96.5%和97.5%, 这表明反应温度的升高有利于苯酚氧化.另外, 由图 5b可以看出, 在200 ℃和2 MPa下, COD去除率随苯酚初始浓度的增加而增加.这主要是因为苯酚初始浓度增加, 使得吸附在催化剂表面的苯酚浓度增加, 从而促进了反应速率的加快.
 |
| 图 5 反应温度(a)和苯酚初始浓度(b)对COD去除率的影响 Fig. 5 Effect of reaction temperature (a) and initial phenol concentrations (b) on the COD removal |
当催化剂在高温条件下催化氧化降解小分子酸时, 金属元素会从固相催化剂中流失到水溶液中.金属元素在溶液中的流失是造成催化剂失活的重要因素之一, 同时金属的溶出也会造成水质的二次污染.因此, 本论文通过ICP-AES测定了经不同催化剂处理后苯酚废水中的金属含量.结果发现, 使用CuO/ZnO/CeO2-ZrO2催化剂催化苯酚废水后的样品, 未检测出Ce和Zr元素, 这主要是由于CeO2-ZrO2载体具有较强的作用力, 以稳定的固溶体形式存在.另外, 使用CuO/ZnO/CeO2和CuO/ZnO/CeO2-ZrO2催化剂催化苯酚废水后的样品, 也未检测出铜元素;使用CuO/ZnO/ZrO2催化剂催化苯酚废水后的样品, 铜的溶出量仅为0.045 mg·L-1, 这主要是由于活性组分铜与载体之间的相互作用力较强, 不易溶出, ICP-AES金属溶出情况与XRD形成铈锆固溶体的结果相符.
4 结论(Conclusions)采用共沉淀法制备了不同载体(CeO2、ZrO2、CeO2-ZrO2)的铜基催化湿式空气氧化苯酚废水催化剂.ZrO2载体主要起到了改善铜的分散, CeO2载体主要起到提供储放氧能力.CeO2-ZrO2复合氧化物载体主要以固溶体形式存在, 既改善了活性组分铜的分散和催化剂的氧化还原性能, 又进一步增加了催化剂的储放氧能力, 因而CuO/ZnO/CeO2-ZrO2催化剂表现出最佳的催化氧化苯酚活性.通过H2-TPR和XPS表明, CuO/ZnO/CeO2-ZrO2催化剂存在表相氧和体相氧两种氧物种, 表相氧的存在有利于苯酚的氧化和降解, 从而增加了催化剂的活性.另外, 由于CuO与ZnO或CeO2-ZrO2载体之间存在较强的作用力, 因此CuO/ZnO/CeO2-ZrO2催化剂没有出现金属溶出现象, 有效防止了金属对水的二次污染.在反应条件为200 ℃和2 MPa空气条件下, COD最高去除率为96.5%.
| [${referVo.labelOrder}] | American Public Health Association (APHA). 2005. Standard methods for the examination of water and wastewater [S]. Washington, DC: American Water Works Association, and Water Environment Federation |
| [${referVo.labelOrder}] | Barbier J, Oliviero L, Renard B, et al. 2005. Role of ceria-support noble metal catalysts (Ru, Pd, Pt) in wet air oxidation of nitrogen and oxygen containing compounds[J]. Top in Catalysis, 33(1): 77–86. |
| [${referVo.labelOrder}] | Bhargava S K, Tardio J, Prasad J, et al. 2006. Wet oxidation and catalytic wet oxidation[J]. Indystrial & Engineering Chemistry Research, 45(4): 1221–1258. |
| [${referVo.labelOrder}] | Chang L, Chen I P, Lin S S. 2005. An assessment of the suitable operating conditions for the CeO2/γ-Al2O3 catalyzed wet air oxidation of phenol[J]. Chemosphere, 58(4): 485–492. DOI:10.1016/j.chemosphere.2004.09.011 |
| [${referVo.labelOrder}] | Chen I P, Lin S S, Wang C H, et al. 2004. Preparing and characterizing an optimal supported ceria catalyst for the catalytic wet air oxidation of phenol[J]. Applied Catalysis B: Environmental, 50(1): 49–58. DOI:10.1016/j.apcatb.2003.12.019 |
| [${referVo.labelOrder}] | Chen I P, Lin S S, Wang C H. 2007. CWAO of phenol using CeO2/γ-Al2O3 with promoter-Effectiveness of promoter addition and catalyst regeneration[J]. Chemosphere, 66(1): 172–178. DOI:10.1016/j.chemosphere.2006.05.023 |
| [${referVo.labelOrder}] | Debellefontaine H, Foussard J N. 2000. Wet air oxidation for the treatment of industrial wastes. Chemical aspects, reactor design and industrial applications in Europe[J]. Waste Management, 20(1): 15–25. DOI:10.1016/S0956-053X(99)00306-2 |
| [${referVo.labelOrder}] | Figueiredo T R, Martinez-Arias A, Granados L M, et al. 1998. Spectroscopic evidence of Cu-Al interactions in Cu-Zn-Al mixed oxide catalysts used in CO hydrogenation[J]. Journal of Catalysis, 178(1): 146–152. DOI:10.1006/jcat.1998.2106 |
| [${referVo.labelOrder}] | Keav S, Monteros A E, Barbier J, et al. 2014. Wet air oxidation of phenol over Pt and Ru catalysts supported on cerium-based oxides: resistance to fouling and kinetic modeling[J]. Applied Catalysis B-Environmental, 150-151: 402–410. DOI:10.1016/j.apcatb.2013.12.028 |
| [${referVo.labelOrder}] | Kim K H, Ihm S K. 2011. Heterogeneous catalytic wet air oxidation of refractory organic pollutant in industrial wastewaters: A review[J]. Journal of Hazardous Materials, 186(1): 16–34. DOI:10.1016/j.jhazmat.2010.11.011 |
| [${referVo.labelOrder}] | Kim K H, Kim J R, Ihm S K. 2009. Wet oxidation of phenol over transition metal oxide catalysts supported on Ce0.65Zr0.35O2 prepared by continuous hydrothermal synthesis in supercritical water[J]. Journal of Hazardous Materials, 167(1): 1158–1162. |
| [${referVo.labelOrder}] | Kim S K, Ihm S K. 2005. Nature of carbonaceous deposits on the alumina supported transition metal oxide catalysts in the wet air oxidation of phenol[J]. Top in Catalysis, 33(1): 171–179. |
| [${referVo.labelOrder}] | Lin S S, Chang D J, Wang C H, et al. 2003. Catalytic wet oxidation of phenol by CeO2 catalyst-effect of reaction conditions[J]. Water Research, 37(4): 793–800. DOI:10.1016/S0043-1354(02)00422-0 |
| [${referVo.labelOrder}] | Lousteau C, Besson M, Descorme C. 2015. Catalytic wet air oxidation of ammonia over supported noble metals[J]. Catalysis Today, 241: 80–85. DOI:10.1016/j.cattod.2014.03.043 |
| [${referVo.labelOrder}] | Monte R D, Kaspar J. 2005. Nanostructured CeO2-ZrO2 mixed oxides[J]. Journal of Materials Chemistry, 15(6): 633–648. DOI:10.1039/B414244F |
| [${referVo.labelOrder}] | Mullins D R, overbury S H, huntley D R. 1998. Electron spectroscopy of single crystal and polycrystalline cerium oxide surfaces[J]. Surface Science, 409(2): 307–319. DOI:10.1016/S0039-6028(98)00257-X |
| [${referVo.labelOrder}] | Oliviero L, Barbier J, Duprez D. 2003. Wet air oxidation of nitrogen-containing organic compounds and ammonia in aqueous media[J]. Applied Catalysis B: Environmental, 40(3): 163–184. DOI:10.1016/S0926-3373(02)00158-3 |
| [${referVo.labelOrder}] | Rocha M A L, Angel G D, Torres-Torres G, et al. 2015. Effect of the Pt oxidation state and Ce3+/Ce4+ ratio on the Pt/TiO2-CeO2 catalysts in the phenol degradation by catalytic wet air oxidation (CWAO)[J]. Catalysis Today, 250: 145–154. DOI:10.1016/j.cattod.2014.09.016 |
| [${referVo.labelOrder}] | Santos A, Yustos P, Durban B, et al. 2001. Catalytic wet oxidation of phenol: kinetics of the mineralization[J]. Indystrial & Engineering Chemistry Research, 40(13): 2773–2781. |
| [${referVo.labelOrder}] | Shimokawabe M, Asakawa H, Takezawa N. 1990. Characterization of copper/zirconia catalysts prepared by an impregnation method[J]. Applied Catalysis, 59(1): 45–58. DOI:10.1016/S0166-9834(00)82186-7 |
| [${referVo.labelOrder}] | 王华, 陶润先, 李光明. 2007. 滴流床反应器中苯酚催化湿式氧化集总反应动力学研究[J]. 环境科学学报, 2007, 27(3): 396–401. |
| [${referVo.labelOrder}] | Wang X H, Lu G Z, Guo Y, et al. 2008. Properties of CeO2-ZrO2 solid solution supported on Si-modified alumina and its application in Pd catalyst for methane combustion[J]. Chinese Journal of Catalysis, 29(10): 1043–1050. DOI:10.1016/S1872-2067(08)60081-9 |
| [${referVo.labelOrder}] | Wu Q, Hu X, Yue P L, et al. 2001. Copper/MCM-41 as catalyst for the wet oxidation of phenol[J]. Applied Catalysis B: Environmental, 32(3): 151–156. DOI:10.1016/S0926-3373(01)00131-X |
| [${referVo.labelOrder}] | Yadav B R, Grag A. 2014. Catalytic wet oxidation of ferulic acid (a lignin model compound) in the presence of non-noble metal based catalysts at mild conditions[J]. Chemical Engineering Journal, 252: 185–193. DOI:10.1016/j.cej.2014.04.110 |
| [${referVo.labelOrder}] | Yang S X, Besson M, Descorme C. 2015. Catalytic wet air oxidation of succinic acid over Ru and Pt catalysts supported on CexZr1-xO2 mixed oxides[J]. Applied Catalysis B: Environmental, 165: 1–9. DOI:10.1016/j.apcatb.2014.09.057 |
| [${referVo.labelOrder}] | Yu Y, Wei H Z, Yu L, et al. 2016. Catalytic wet air oxidation of m-cresol over a surface-modified sewage sludge-derived carbonaceous catalyst[J]. Catalysis Science & Technology, 6(4): 1085–1093. |
| [${referVo.labelOrder}] | Zhang L, Pan L W, Ni C J, et al. 2013a. Effect of precipitation aging time on the performance of CuO/ZnO/CeO2-ZrO2 for methanol steam reforming[J]. Journal of Fuel Chemistry and Technology, 41(7): 883–888. DOI:10.1016/S1872-5813(13)60038-9 |
| [${referVo.labelOrder}] | Zhang L, Pan L W, Ni C J, et al. 2013b. CeO2-ZrO2-promoted CuO/ZnO catalyst for methanol steam reforming[J]. International Journal of Hydrogen Energy, 38(1): 4397–4406. |
| [${referVo.labelOrder}] | 张磊. 2013. 甲醇水蒸汽重整制氢催化剂的研究[D]. 大连: 大连理工大学. 18-22 |
| [${referVo.labelOrder}] | 张磊, 雷俊腾, 田园, 等. 2015. 前驱体和沉淀剂浓度对CuO/ZnO/CeO2-ZrO2甲醇水蒸气重整制氢催化剂性能的影响[J]. 燃料化学学报, 2015, 43(11): 1366–1374. DOI:10.3969/j.issn.0253-2409.2015.11.012 |
| [${referVo.labelOrder}] | 张磊, 潘立卫, 倪长军, 等. 2012. 沉淀温度对CuO/ZnO/CeO2/ZrO2甲醇水蒸气重整制氢催化剂性能的影响[J]. 催化学报, 2012, 33(12): 1958–1964. |