 2017, Vol. 37
2017, Vol. 37



森林植被释放巨量生物源挥发性有机化合物(biogenic volatile organic compounds, BVOC), 它们和大气中的氧化性物种, 如OH自由基、O3、NO3自由基等, 发生光氧化反应, 生成低挥发性的极性有机化合物, 经气-粒转化, 形成二次有机气溶胶(secondary organic aerosols, SOA), 是PM2.5的重要组分, 对空气质量、全球气候和人体健康影响极大.其中, 异戊二烯全球年排放量为309~706 Tg, 占BVOC的50%(Navarro et al., 2014; Guenther et al., 2006), 其光氧化产物, 2-甲基丁四醇(2-methyltetrols, 2-MT)、2-甲基甘油酸(2-methylglyceric acid, 2-MG)(Claeys et al., 2004a; Edney et al., 2005; Claeys et al., 2004b), 在森林和植被丰富的城市地区PM2.5中, 被相继发现, 且具有相当的浓度, 是异戊二烯SOA的示踪物.α-蒎烯是BVOC中排放量最大的单萜类化合物, 全球年排放量为89 Tg(Navarro et al., 2014), 3-甲基-1, 2, 3-丁基三羧酸(3-methyl-1, 2, 3-butanetricarboxylic acid, MBTCA)是α-蒎烯的光氧化产物(Jaoui et al., 2005), 也是α-蒎烯SOA的示踪物之一(Szmigielski et al., 2007).真菌孢子作为天然源一次颗粒可直接排放到大气中(Després et al., 2012), 或通过土壤再悬浮排放到大气中(Fröhlich Nowoisky et al., 2012), 阿拉伯糖醇(arabitol)、甘露糖醇(mannitol)可作为真菌孢子的示踪物(Bauer et al., 2008).
以甲苯为代表的人为源VOC, 主要以机动车尾气和工业排放的芳香族化合物为主(Volkamer et al., 2001).中国年排放量达到23 Tg(Zhang et al., 2009).2, 3-二羟基-4-氧代戊酸(2, 3-dihydroxy-4-oxopentanoic acid, DHOPA)为人为源芳香烃经光氧化所产生的氧化产物(Kleindienst et al., 2004).另外, 生物质燃烧过程中, 纤维素经热解产生左旋葡聚糖(levoglucosan), 半纤维素经热解生成半乳聚糖(galacopyranose)和甘露聚糖(mannopyranose), 均为强极性化合物(Velasco et al., 2007), 由于左旋葡聚糖的特异性、光化学稳定性以及释放量大的特点, 使其被公认且被广泛的用作生物质燃烧的示踪物(Engling et al., 2009).城市地区大气环境受人为因素影响较为严重, 因此, 在人为活动较为密集的城市地区, 大气环境更加复杂多变, 对城市地区气溶胶的成分组成、时空分布及影响因素的研究也有很大意义.
本文在金华市内的郊区(Suburban)、城区(Urban)、工业区(Industrial)设立采样点, 采集了四个季节, 共225个PM2.5样品, 通过气相色谱/质谱(GC/MS), 分析了极性有机成分, 对以异戊二烯和α-蒎烯为代表的生物源SOA, 和以甲苯为代表的人为源SOA示踪物, 以及生物质燃烧示踪物和阿拉伯糖醇、甘露糖醇的浓度水平、季节变化和时空分布进行了研究, 并对SOA及其它组分对有机碳的贡献进行了估算.以期了解金华地区气溶胶中极性有机化合物的组成状况, 并对金华市大气环境质量和污染防治提供科学依据.
2 采样和分析(Samples and analysis) 2.1 采样点描述采样地点位于浙江省金华市, 3个采样点分别为位于金东区的环境监测中心(119°41′48″E, 29°6′21″N)、开发区的第十五中学(119°39′33″E, 29°4′49″N)以及婺城区的婺城小学(119°34′19″E, 29°5′47″N), 如图 1所示.其中, 环境监测中心位于金华市区东部, 属于新开发区域, 建筑密度、交通密度和工业活动强度较小, 列为郊区(Suburban)站点; 第十五中学地处市中心区, 以居住和办公区为主, 建筑密集, 交通流量较大, 为市区(Urban)站点; 婺城小学位于金华市西部, 受工业活动的影响较大, 属于工业区(Industrial)站点.采样时间为2015年, 四个季节采样日期分别为:1月20-2月7日(冬季); 4月11-4月30日(春季); 7月16-8月4日(夏季); 10月15-11月4日(秋季).采样期间用大流量采样器(GUV16HBL, Thermo-Andersen, USA, 1000 L·min-1)24 h连续采样, 每天采集1个PM2.5样品, 包括温度和相对湿度数据.
 |
| 图 1 采样点位置示意图 Fig. 1 Location of the sampling sites |
所用标准试剂有左旋葡聚糖(1, 6-anhydro-β-glucopyranose, 98%, Fluka)、半乳聚糖(1, 6-anhydro-β-D-galacopyranose, 98%, Sigma)、甘露聚糖(1, 6-anhydro-β-D-mannopyranose, 98%, Sigma)、阿拉伯糖醇(L-(-)-arabitol, 99%, Fluka)、甘露糖醇(D(-)-mannitol, 98%, Merck)、赤藓糖醇(meso-erythritol, 99%, Fluka)、苹果酸(D(+)-malic acid, 97%, Fluka)、1, 4-环己烷二甲酸(1, 4-cyclohexanedicarboxylic acid, 99%, Aldrich)、2, 3-二羟基丁二酸(DL-2, 3-dihydroxybutanedioic acid, 99%, Sigma)、甲基-β-D-吡喃木糖苷(methyl-β-D-xylanopyranoside, 99%, Sigma); 甲醇(methanol, 99.9%, CNW)、二氯甲烷(dichloromethane, 99.9%, CNW).
安捷伦(Agilent, USA)气相色谱-质谱联用仪(6890/5975, GC-MS), DB-5MS(30 m×0.25 mm×0.25 μm)毛细管色谱柱, 载气为高纯氦气, 流速为1.0 mL·min-1.离子源为EI, 电子能量为70 eV.升温程序为初始温度70 ℃, 保持2 min, 以3.2 ℃·min-1的速率升至200 ℃, 保持5 min, 再以5 ℃·min-1的速率升至300 ℃, 最后在300 ℃下后运行5 min.
2.3 样品分析取样品3 cm2, 加适量内标MXP(methyl-β-D-xylanopyranoside), 用二氯甲烷/甲醇(1 : 1, V/V)超声抽提, 合并提取液, 浓缩、吹干.BSTFA (N, O-bis(trimethylsilyl)-Trifluoroacetamide)+1%TMS衍生, GC/MS分析.根据文献识别并定量了5种SOA示踪物, 包括异戊二烯SOA示踪物:2-甲基甘油酸(2-MG)(Claeys et al., 2004b)、2-甲基丁四醇(2-MT):2-甲基赤藓糖醇(2-methylerythritol)和2-甲基苏糖醇(2-methylthreitol) (Claeys et al., 2004a); α-蒎烯SOA示踪物:3-甲基-1, 2, 3-丁基三羧酸(MBTCA)(Jaoui et al., 2005); 甲苯SOA示踪物:2, 3-二羟基-4-氧代戊酸(DHOPA)(Kleindienst et al., 2004); 3种脱水糖类化合物(Simoneit et al., 1999), 包括生物质燃烧示踪物左旋葡聚糖, 及其同分异构体半乳聚糖和甘露聚糖; 以及2种糖醇类化合物, 阿拉伯糖醇和甘露糖醇(Simoneit et al., 2004).其中2种糖醇和3种脱水糖都用标准物质定量, 2-甲基赤藓糖醇和2-甲基苏糖醇用赤藓糖醇定量(Wang et al., 2008), 2-甲基甘油酸用苹果酸定量(Edney et al., 2005), 3-甲基-1, 2, 3-丁基三羧酸用1, 4-环己烷二甲酸定量(Wang et al., 2008), 2, 3-二羟基-4-氧代戊酸用2, 3-二羟基丁二酸定量.
2.4 质量保证与控制每个季节、每个采样点都采集了1个空白样, 样品预处理的过程中每次处理1批样品都加入1个实验空白, 以检验实验操作过程中有无杂质带入.样品空白和实验空白均无影响目标化合物的杂质出现.仪器分析过程中, 每进10个样品进1个重复样, 每进20个样品后要进1个标样, 检验仪器状态是否正常, 两次平行样检测结果的差值应小于20%, 确保仪器状态的稳定.
2.5 二次有机碳(SOC)估算方法用示踪物-产率法进行SOC的估算, 其原理为利用烟雾箱实验模拟各VOC产生SOA的过程, 通过GC/MS测定每种VOC前体物在烟雾箱中反应生成的一系列有机示踪物的浓度, 将所有SOA有机示踪物的浓度之和, 除以烟雾箱反应后产生的总有机碳(即总SOC)的浓度, 即为SOC的质量分数(FSOC), 通过测定实际大气中各种前体物产生的SOA示踪物浓度, 借助FSOC可估算某一特定VOC前体物产生的SOC浓度, 并推算不同来源SOC对OC的贡献(Kleindienst et al., 2007).
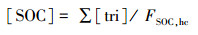
|
(1) |
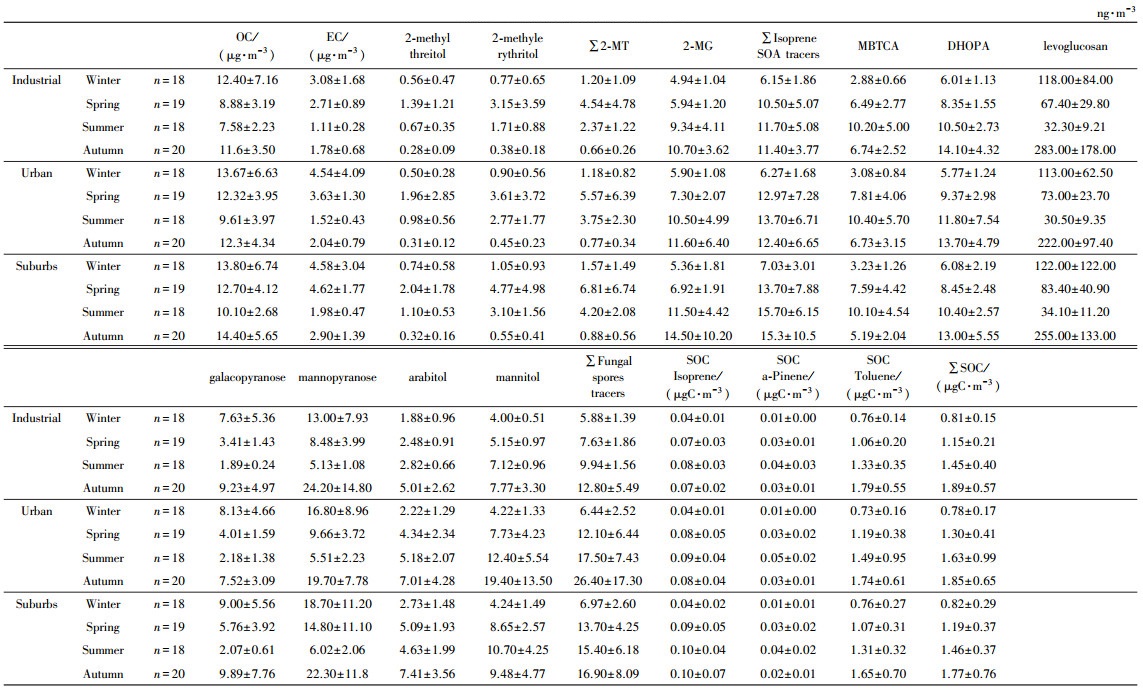
3个采样点异戊二烯SOA示踪物浓度范围分别为:郊区3.85~25.90 ng·m-3, 平均值为9.94 ng·m-3; 市区3.41~33.90 ng·m-3, 平均值为11.50 ng·m-3; 工业区3.41~48.50 ng·m-3, 平均值为13.00 ng·m-3.如表 1所示为各SOA示踪物的浓度分布.3个采样点之间有较为明显的变化, 可表示为工业区 > 市区 > 郊区.如图 3所示为3个采样点各SOA示踪物浓度的季节变化.从图中可以看出, SOA示踪物浓度的季节变化非常明显.其中, 异戊二烯SOA示踪物浓度夏季最高, 冬季最低, 各季节的平均浓度分别为冬季6.42 ng·m-3, 春季12.40 ng·m-3, 夏季13.70 ng·m-3, 秋季13.00 ng·m-3, 研究表明异戊二烯的排放速率与温度和光照成正相关性(Rinne et al., 2002), 而夏季的强光照和高温也有利于大气中异戊二烯的光氧化反应(Volkamer et al., 2001), 因此, 受季节、温度和光照变化对异戊二烯排放和大气氧化环境的影响, 造成夏季浓度最高冬季浓度最低的变化趋势, 与广州、上海等地的相关研究一致(Ding et al., 2012; Feng et al., 2013).此外, 气候、季风、气象条件、地理位置等对SOA示踪物浓度也有很大影响, 例如在印度孟买由于受西南方向的夏季风带来清洁的海上气团的影响, 天然源SOA示踪物浓度在夏季要比冬季低几倍(Fu et al., 2016).
| 表 1 各组分浓度水平 Table 1 Concentrations of the components |
 |
| 图 3 各SOA示踪物浓度和总SOC的季节变化 Fig. 3 Seasonal variations of concentration of SOA tracers and total SOC |
异戊二烯示踪物包括2-甲基丁四醇和2-甲基甘油酸.其中2-甲基丁四醇浓度范围为0.13~26.00 ng·m-3, 平均浓度为2.75 ng·m-3, 2-甲基甘油酸浓度为3.20~47.50 ng·m-3, 平均浓度为8.74 ng·m-3, 2-甲基甘油酸浓度明显高于2-甲基丁四醇.相关研究表明(Surratt et al., 2010; Paulot et al., 2009; Lin et al., 2013), 2-甲基丁四醇和2-甲基甘油酸分别由不同路径产生:HO2-channel和NOx-channel, 两者共同存在且相互竞争.2-甲基丁四醇经HO2-channel路径生成.在人为活动排放的影响下, NOx(NO+NO2)浓度的升高使得异戊二烯SOA形成路径由HO2-channel转向NOx-channel, 生成2-甲基甘油酸.两条路径的变化, 取决于异戊二烯和NOx的相对浓度, 而两者随地点和季节变化而各有不同.在高NOx浓度下, 异戊二烯的主要氧化产物是2-甲基甘油酸(Carlton et al., 2009), 本研究与长白山自然保护区(Wang et al., 2008)相比, 2-甲基甘油酸的浓度明显高于长白山森林地区, 印证了金华地区人为活动多, 大气中较高浓度的NOx(NO2体积分数为金华13~27 ppbv, 长白山0~6.4 ppbv), 使得异戊二烯光氧化反应由NOx-channel主导, 因此有更多2-甲基甘油酸产生.
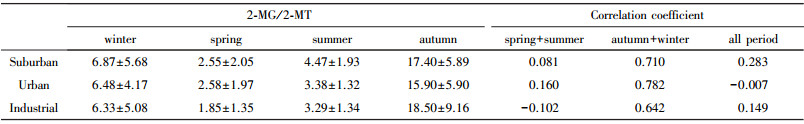
2-甲基甘油酸/2-甲基丁四醇(2-MG/2-MT)的比值可以反映NOx对异戊二烯SOA形成的影响(Ding et al., 2013).烟雾箱实验证明, 随着NOx/异戊二烯比值升高, 2-MG/2-MT的比值也升高(Edney et al., 2005; Jaoui et al., 2010; Surratt et al., 2007).因此, 2-MG/2-MT比值可以反应NOx对异戊二烯SOA形成的影响, 即若不同地区异戊二烯浓度差异不大, 则NOx浓度越高, 2-MG/2-MT比值越高.表 2为各采样点不同季节的2-MG/2-MT比值, 各季节比值最高的站点分别为:冬季郊区最高、春季城区最高、夏季郊区最高、秋季工业区最高, 不同采样点之间受季节变化影响较大.Ding等(2016)通过对生物质燃烧示踪物左旋葡聚糖的研究发现, 中国秋季、冬季到春季(10月—4月), 野外秸秆等生物质燃烧从重到轻, 而生物质燃烧可释放大量的异戊二烯和NOx, 从而对异戊二烯SOA有很重要的影响.本文2-MG/2-MT比值在各采样点不同季节之间有较为明显的差异, 表现为秋季最高, 冬季次之, 而春季和夏季较低; 秋、冬季较高的2-MG/2-MT比值与秋冬季节较多的生物质燃烧相符.为进一步验证, 我们对不同季节的异戊二烯示踪物和生物质燃烧示踪物(左旋葡聚糖)作相关性分析, 如表 2所示, 在春、夏季以及全年的分析中两者均没有明显相关性, 而秋、冬季两者则具有显著相关性(p < 0.01).
| 表 2 各采样点不同季节的2-MG/2-MT比值以及异戊二烯SOA示踪物与左旋葡聚糖的相关性 Table 2 Ratio of 2-MG/2-MT and correlations between isoprene SOA tracers and levoglucsan at different sampling sites during different sampling periods |
α-蒎烯SOA示踪物在3个采样点的浓度为:郊区为2.64~20.9 ng·m-3, 平均值为6.58 ng·m-3, 市区为2.48~25.40 ng·m-3, 平均值为6.85 ng·m-3, 工业区为2.45~23.20 ng·m-3, 平均值为6.53 ng·m-3.3个采样点之间浓度水平非常接近, 没有明显变化.各季节平均浓度分别为冬季3.06 ng·m-3, 春季7.29 ng·m-3, 夏季10.30 ng·m-3, 秋季6.22 ng·m-3, 可以看出不同季节之间浓度变化非常明显, 如图 3所示, 表现为夏季>春季≈秋季>冬季, 主要是由于季节变化对α-蒎烯排放的影响, 以及夏季的高温、强光照, 更有利于3-甲基-1, 2, 3-丁基三羧酸的生成(Feng et al., 2013).3-甲基-1, 2, 3-丁基三羧酸在本研究中作为主要的α-蒎烯SOA示踪物, 与广州地区(Ding et al., 2012)较为接近, 稍高于泰山地区, 主要由于泰山地处北温带, 植被为落叶阔叶林, 且采样点山顶多为灌木、地面多岩石(Fu et al., 2010).本研究夏季3-甲基-1, 2, 3-丁基三羧酸的浓度(3.62~25.40 ng·m-3)明显低于香港地区相同季节数据(1.69~457.00 ng·m-3), 虽然同为城市地区, 但由于香港地区城市绿化面积大、植被覆盖面积广、植被种类多, 使得香港地区有较高的α-蒎烯排放, 产生较高浓度的3-甲基-1, 2, 3-丁基三羧酸(Hu et al., 2006).
3.1.3 真菌孢子示踪物阿拉伯糖醇和甘露糖醇两种糖醇, 作为真菌孢子的示踪物, 在金华地区的浓度为:阿拉伯糖醇0.76~16.40 ng·m-3, 平均值为4.23 ng·m-3; 甘露糖醇3.03~45.70 ng·m-3, 平均值为10.80 ng·m-3.远低于植物和微生物活动密集的森林地区(阿拉伯糖醇(156±102) ng·m-3, 甘露糖醇(349±243) ng·m-3)(Zhu et al., 2016), 而高于相当于地球大气真菌孢子含量背景值的沙漠地区(阿拉伯糖醇0.11~5.60 ng·m-3, 甘露糖醇0.02~1.90 ng·m-3)(Fu et al., 2016), 与北京地区较为接近(阿拉伯糖醇0.70~74.20 ng·m-3, 平均值为7.40 ng·m-3, 甘露糖醇0.90~95.30 ng·m-3, 平均值为10.30 ng·m-3)(Liang et al., 2016).如表 1所示为具体浓度分布, 从季节分布来看, 3个采样点均为冬季浓度最低, 可能的原因是冬季低温、干燥、植物和微生物活动减少等.
如图 2所示, 在本研究中, 阿拉伯糖醇、甘露糖醇均与温度呈正相关性, R2分别为0.67和0.70, 表明温度升高有利于真菌孢子的形成.Zhu等(2016)在研究中发现高湿度的环境有利于真菌孢子的形成, 而本文中真菌孢子示踪物与湿度无明显正负相关性, R2分别为0.34和0.07.不同种类的真菌排放到大气中的真菌孢子有很大不同, 取决于地理位置、季节、天气以及一天之中的时间等, 不同种类的真菌孢子可能包含不同浓度水平的阿拉伯糖醇和甘露糖醇(Yang et al., 2016).本研究中甘露糖醇浓度和阿拉伯糖醇浓度的比值分别为:郊区0.70~ 4.94, 平均值为2.32, 市区0.88~4.29, 平均值为2.15, 工业区0.86~3.96, 平均值为1.84.且阿拉伯糖醇和甘露糖醇在3个采样点均具有显著相关性(p < 0.01), 相关系数分别为郊区0.776, 市区0.892, 工业区0.795, 表明金华地区真菌孢子的来源较为稳定、真菌种类较为固定.
 |
| 图 2 阿拉伯糖醇、甘露糖醇和温度的散点图 Fig. 2 Scatter plots of arabitol, mannitol and temperature |
从全球范围来看, 天然源VOCs在大气环境中占据主导地位(Guenther et al., 2006), 而在人口密度大, 工业化程度高的城市地区, 人为源VOCs对大气环境具有重要影响.本研究中甲苯SOA示踪物的浓度范围为4.75~39.80 ng·m-3, 与北京(Guo et al., 2012)(1.00~42.00 ng·m-3)浓度相近; 高于上海(Feng et al., 2013)(0.04~10.0 ng·m-3), 该研究中所有SOA示踪物均使用顺式-蒎酮酸(KPA)定量, 而本文使用2, 3-二羟基丁二酸对2, 3-二羟基-4-氧代戊酸进行定量; 与广州(Ding et al., 2012)(2.84~52.0 ng·m-3)相比较低, 该研究所报道的甲苯SOA示踪物浓度在近年来相关研究中都属于较高水平, 而珠三角地区人为源VOCs(甲苯、二甲苯)的排放量也属于较高水平(Chan et al., 2006; Barletta et al., 2008), 加之接近热带的气候环境, 因此有较高的甲苯SOA示踪物浓度.甲苯SOA示踪物浓度各季节平均浓度分别为冬季5.94 ng·m-3、春季8.70 ng·m-3、夏季10.9 ng·m-3、秋季13.6 ng·m-3, 有较为明显的季节变化, 如图 3所示, 表现为秋季 > 夏季 > 春季 > 冬季, 此变化趋势与上海(Feng et al., 2013)相似.而根据相关研究(Huang et al., 2011; Liu et al., 2010), 长江三角洲地区人为源芳香烃VOCs排放多来源于化工厂、涂漆装修以及汽车尾气排放等, 季节变化相对较小, 本研究中夏秋季节的甲苯SOA示踪物的高浓度可能是由于夏秋季节的高温、强光照所导致的强氧化作用以及秋季特殊气象条件所导致的污染物积累.甲苯SOA示踪物在3个采样点之间的浓度分布为:郊区5.14~20.20 ng·m-3, 平均值为9.72 ng·m-3, 市区4.75~39.80 ng·m-3, 平均值为10.40 ng·m-3, 工业区4.75~22.70 ng·m-3, 平均值为9.53 ng·m-3.可以看出市区平均浓度稍高于工业区和郊区, 而且浓度变化范围也更大, 这说明由于市区人口密度大、机动车排放等人为活动更多, 对人为源VOCs的排放有很大影响.实际观测研究(Barletta et al., 2005; Zhang et al., 2013)以及排放清单研究(Zhang et al., 2009; Zheng et al., 2009)结果显示中国的人为源芳香烃主要来源于有机溶剂的使用、化石燃料燃烧(包括工业、机动车等), 以及生物质燃烧.有机溶剂的使用和化石燃料燃烧在城市地区对人为源芳香烃的贡献更高, 而生物质燃烧(森林大火、生物燃料燃烧、作物残渣露天燃烧)在郊区对人为源芳香烃具有较高的贡献(Ding et al., 2014), 因此可能由于受生物质燃烧的影响在郊区也能检测到较高浓度的2, 3-二羟基-4-氧代戊酸.
由于SOA是由前体物VOC在大气中发生光氧化反应生成的, 在对SOA示踪物浓度与大气中O3、NO2、SO2浓度(数据来自与中国环境监测总站)进行拟合时也发现, 夏季各示踪物浓度以及总SOC浓度都与臭氧浓度具有明显的正相关性, 如图 4、5所示, 异戊二烯R2=0.733、α-蒎烯R2=0.629、甲苯R2=0.694、总SOC R2=0.620, 表明O3对SOC的形成有直接影响.
 |
| 图 4 各SOA示踪物与臭氧的散点图 Fig. 4 Scatter plots of SOA tracers and O3 |
 |
| 图 5 总SOC与臭氧的散点图 Fig. 5 Scatter plots of total SOC and O3 |
生物质燃烧示踪物浓度水平如表 1所示, 其中左旋葡聚糖浓度范围为郊区16.8~732.0 ng·m-3, 平均值为131.0 ng·m-3, 市区19.1~491.0 ng·m-3, 平均值为114.0 ng·m-3, 工业区19.1~657.0 ng·m-3, 平均值为128.0 ng·m-3, 是本研究中浓度水平最高的有机化合物, 且高于北京地区(34.0~149.0 ng·m-3)(Yang et al., 2016).高浓度的左旋葡聚糖水平说明, 生物质燃烧排放对金华地区有机气溶胶具有重要贡献.而且左旋葡聚糖浓度有明显的季节变化, 如图 6所示, 冬季和秋季左旋葡聚糖浓度高、浓度变化幅度大, 可能是因为秋冬季节生物质燃烧量增多所导致.半乳聚糖和甘露聚糖是半纤维素的热解产物, 而左旋葡聚糖是纤维素的热解产物(Velasco et al., 2007).而硬木含有的纤维比半纤维多, 因此左旋葡聚糖/甘露聚糖(L/M)的比值可以用来区分硬木(被子植物)和软木(裸子植物)的燃烧(Verma et al., 2015).相关研究报道, 作物残渣燃烧L/M比值> 40, 硬木燃烧为15~25, 软木燃烧为3~10(Schmidl et al., 2008; Ding et al., 2012; Mkoma et al., 2013).本研究中, L/M比值为1~23, 因此, 金华地区主要生物质燃烧源为草本植物和硬木.
 |
| 图 6 左旋葡聚糖浓度的季节变化 Fig. 6 Seasonal variations of levoglucosan concentration |
利用换算因子(OC/levoglucosan的比值)可以估算气溶胶中来源于生物质燃烧的OC(OC生物质燃烧)浓度, 换算因子根据燃烧环境和植被种类的不同而变化, 因子越小越趋近于软木燃烧, 越高越趋近于硬木燃烧(Puxbaum et al., 2007).Zhang等(2007)报道该因子在中国范围为8~16, 这里设转换因子为10.各采样点OC生物质燃烧浓度分别为:郊区0.17~7.32 μg·m-3, 平均值1.26 μg·m-3; 市区0.18~4.91 μg·m-3, 平均值1.14 μg·m-3; 工业区0.19~6.57 μg·m-3, 平均值1.28 μg·m-3.对OC的贡献分别可以达到:郊区3.18%~42.8%, 市区1.44%~28.0%, 工业区1.23%~26.70%, 可以看出3个采样点中, 郊区受生物质燃烧的影响最大, 可能由于郊区更多的生物燃料燃烧以及作物残渣露天燃烧等现象所致.不同季节OC生物质燃烧浓度分别为:冬季0.17~5.79 μg·m-3, 平均值为1.18 μg·m-3; 春季0.34~2.05 μg·m-3, 平均值为0.75 μg·m-3; 夏季0.18~0.64 μg·m-3, 平均值为0.32 μg·m-3; 秋季0.82~7.32 μg·m-3, 平均值为2.53 μg·m-3.有明显的季节变化, 表现为秋季>冬季>春季>夏季, 对OC的贡献平均值依次为秋季19.5%, 冬季8.69%, 春季6.93%, 夏季3.80%.Feng等(2013)对上海地区的估算结果中生物质燃烧对水溶性有机碳的贡献可达到54%, 且与本研究具有相同的季节变化趋势.
3.3 SOC估算及其对OC的贡献Kleindienst等(2007)通过烟雾箱实验模拟VOCs产生SOA的过程, 建立了一种利用大气中可检测的VOCs氧化产物作为二次有机示踪物来估算人为源及天然源SOA的方法, 即示踪物-产率法.这种方法已被多次应用于不同地区SOA的估算, John等(2006)对北加利福尼亚地区天然源和人为源VOCs产生的SOA进行了研究, 结果显示4种SOA前体物产生的二次有机碳(SOC)对OC的贡献达到55%, 并发现SOC对OC的贡献与温度有关, 温度最高时SOC对OC的贡献最高, 温度最低时SOC对OC的贡献也最低.Ding等(2012)对中国珠三角地区不同季节的SOC进行了估算, 秋冬季SOC对OC的贡献为8.7%, 而夏季可达到38.4%.Feng等(2013)研究了上海地区SOA的来源和季节变化, 结果显示夏季各SOA示踪物浓度明显高于其他季节, 该研究同时还比较和评价了用不同方法估算SOC的结果.Guo等(2012)研究了北京城市和农村地区的SOA的组成, 城市地区SOC对OC的贡献为32.5%, 农村地区为38.4%, 其中分别有17.4%和17.2%来自于甲苯, 由此得出人为源SOA对北京的空气质量具有重要影响.
利用该方法对SOC进行了估算, 总SOC浓度为0.63~5.19 μgC·m-3, 其中SOC异戊二烯为0.02~0.31 μgC·m-3, SOCα-蒎烯为0.01~0.11 μgC·m-3, SOC甲苯为0.60~5.04 μgC·m-3, 与邻近的上海地区(Feng et al., 2013)相比, SOC异戊二烯和SOCα-蒎烯浓度相近, SOC甲苯浓度高于上海.各SOC都具有明显的季节变化, 表现为夏季最高, 冬季最低, 与各SOA示踪物浓度的季节变化一致.SOC对OC的贡献达到3.34%~26.70%, 在高度工业化和城市化的广州地区, SOC对OC的贡献可达到79.00%(Ding et al., 2012).SOC甲苯对OC的贡献为3.03%~24.50%, SOC异戊二烯对OC的贡献为0.17%~2.23%, SOCα-蒎烯对OC的贡献为0.03%~0.83%.3个采样点之间SOC甲苯对OC的贡献分别为:郊区3.36%~24.50%, 平均值为13.70%;市区3.43%~23.70%, 平均值为11.80%;工业区3.03%~20.10%, 平均值为10.20%, 可以看出SOC甲苯在3个采样点对OC均有较高的贡献, 是SOC的重要组分部分, 表明人为源排放对区域大气环境有重要影响.
4 结论(Conclusions)1) 各SOA示踪物均有明显的季节变化, 主要表现为异戊二烯SOA示踪物:夏季>秋季≈春季>冬季, α-蒎烯SOA示踪物:夏季>春季≈秋季>冬季, 甲苯SOA示踪物:秋季>夏季>春季>冬季.2-甲基甘油酸是主要的异戊二烯SOA示踪物, 且本研究结果验证了生物质燃烧对异戊二烯SOA具有重要的影响.
2) 真菌孢子示踪物的季节变化较为明显, 3个采样点均为冬季浓度最低.阿拉伯糖醇、甘露糖醇均与温度呈正相关性, 表明温度升高有利于真菌孢子的形成.
3) 左旋葡聚糖是本研究中浓度水平最高的有机化合物, 根据左旋葡聚糖和甘露聚糖的比值得出金华地区主要的生物质燃烧来源为草本植物和硬木.
4) 通过估算, 二次有机碳对OC的贡献达到3.34%~26.70%, 其中SOC甲苯对OC具有重要贡献(3.03%~24.50%); 来源于生物质燃烧的有机碳对OC的贡献可以达到1.23%~42.80%, 可以看出人为源排放以及生物质燃烧对金华地区大气环境具有重要影响.
Barletta B, Meinardi S, Rowland F S, et al. 2005. Volatile organic compounds in 43 Chinese cities[J]. Atmospheric Environment, 39(32): 5979–5990.
DOI:10.1016/j.atmosenv.2005.06.029
|
Barletta B, Meinardi S, Simpson I J, et al. 2008. Ambient mixing ratios of nonmethane hydrocarbons (NMHCs) in two major urban centers of the Pearl River Delta (PRD) region: Guangzhou and Dongguan[J]. Atmospheric Environment, 42(18): 4393–4408.
DOI:10.1016/j.atmosenv.2008.01.028
|
Bauer H, Claeys M, Vermeylen R, et al. 2008. Arabitol and mannitol as tracers for the quantification of airborne fungal spores[J]. Atmospheric Environment, 42(3): 588–593.
DOI:10.1016/j.atmosenv.2007.10.013
|
Carlton A G, Wiedinmyer C, Kroll J H, et al. 2009. A review of secondary organic aerosol (SOA) formation from isoprene[J]. Atmospheric Chemistry and Physics, 9(2): 4987–5005.
|
Chan L Y, Chu K W, Zou S C, et al. 2006. Characteristics of nonmethane hydrocarbons (NMHCs) in industrial, industrial-urban, and industrial-suburban atmospheres of the Pearl River Delta (PRD) region of south China[J]. Journal of Geophysical Research, 111(D11): 1937–1952.
|
Claeys M, Graham B, Vas G, et al. 2004a. Formation of secondary organic aerosols through photooxidation of isoprene[J]. Science, 303(5661): 1173–1176.
DOI:10.1126/science.1092805
|
Claeys M, Wang W, Ion A C, et al. 2004b. Formation of secondary organic aerosol from isoprene and its gas-phase oxidation products through reaction with hydrogen peroxide[J]. Atmospheric Environment, 38(25): 4093–4098.
DOI:10.1016/j.atmosenv.2004.06.001
|
Després V R, Huffman J A, Burrows S M, et al. 2012. Primary biological aerosol particles in the atmosphere: a review[J]. Tellus B, 64: 15598.
DOI:10.3402/tellusb.v64i0.15598
|
Ding X, He Q F, Shen R Q, et al. 2014. Spatial distributions of secondary organic aerosols from isoprene, monoterpenes, β-caryophyllene, and aromatics over China during summer[J]. Journal of Geophysical Research, 119(20): 11877–11891.
|
Ding X, He Q F, Shen R Q, et al. 2016. Spatial and seasonal variations of isoprene secondary organic aerosol in China: Significant impact of biomass burning during winter[J]. Scientific Reports, 6: 20411.
DOI:10.1038/srep20411
|
Ding X, Wang X M, Gao B, et al. 2012. Tracer-based estimation of secondary organic carbon in the Pearl River Delta, south China[J]. Journal of Geophysical Research, 117: D05313.
|
Ding X, Wang X M, Xie Z Q, et al. 2013. Impacts of Siberian biomass burning on organic aerosols over the North Pacific Ocean and the Arctic: Primary and secondary organic tracers[J]. Environmental Science & Technology, 47(7): 3149–3157.
|
Edney E O, Kleindienst T E, Jaoui M, et al. 2005. Formation of 2-methyltetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOx/SO2/air mixtures and their detection in ambient PM2.5 samples collected in the eastern United States[J]. Atmospheric Environment, 39(29): 5281–5289.
DOI:10.1016/j.atmosenv.2005.05.031
|
Engling G, Lee J J, Tsai Y W, et al. 2009. Size-resolved anhydrosugar composition in smoke aerosol from controlled field burning of rice straw[J]. Aerosol Science and Technology, 43(7): 662–672.
DOI:10.1080/02786820902825113
|
Feng J L, Li M, Zhang P, et al. 2013. Investigation of the sources and seasonal variations of secondary organic aerosols in PM2.5 in Shanghai with organic tracers[J]. Atmospheric Environment, 79: 614–622.
DOI:10.1016/j.atmosenv.2013.07.022
|
Fröhlich Nowoisky J, Burrows S M, Xie Z, et al. 2012. Biogeopraphy in the air: fungal diversity over land and oceans[J]. Biogeosciences, 9(3): 1125–1136.
DOI:10.5194/bg-9-1125-2012
|
Fu P Q, Kawamura K, Kanaya Y, et al. 2010. Contributions of biogenic volatile organic compounds to the formation of secondary organic aerosols over Mt.Tai, Central East China[J]. Atmospheric Environment, 44(38): 4817–4826.
DOI:10.1016/j.atmosenv.2010.08.040
|
Fu P Q, Zhuang G S, Sun Y L, et al. 2016. Molecular markers of biomass burning, fungal spores and biogenic SOA in the Taklimakan desert aerosols[J]. Atmospheric Environment, 130: 64–73.
DOI:10.1016/j.atmosenv.2015.10.087
|
Guenther A, Karl T, Harley P, et al. 2006. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature)[J]. Atmospheric Chemistry and Physics, 6: 3181–3210.
DOI:10.5194/acp-6-3181-2006
|
Guo S, Hu M, Guo Q F, et al. 2012. Primary Sources and Secondary Formation of Organic Aerosols in Beijing, China[J]. Environmental Science & Technology, 46(18): 9846–9853.
|
Huang C, Chen C H, Li L, et al. 2011. Emission inventory of anthropogenic air pollutants and VOC species in the Yangtze River Delta region, China[J]. Atmospheric Chemistry and Physics, 11(9): 4105–4120.
DOI:10.5194/acp-11-4105-2011
|
Hu D, Bian Q J, Li T W Y, et al. 2008. Contributions of isoprene, monoterpenes, b-caryophyllene, and toluene to secondary organic aerosols in Hong Kong during the summer of 2006[J]. Journal of Geophysical Research, 113: D22206.
DOI:10.1029/2008JD010437
|
Jaoui M, Kleindienst T E, Lewandowski M, et al. 2005. Identification and quantification of aerosol polar oxygenated compounds bearing carboxylic or hydroxyl groups 2. Organic tracer compounds from monoterpenes[J]. Environmental Science & Technology, 39(15): 5661–5673.
|
Jaoui M, Corse E W, Lewandowski M, et al. 2010. Formation of organic tracers for isoprene SOA under acidic conditions[J]. Atmospheric Environment, 44(14): 1798–1805.
DOI:10.1016/j.atmosenv.2010.01.018
|
John H O, Michael L, Mohammed J, et al. 2011. Contributions of Biogenic and Anthropogenic Hydrocarbons to Secondary Organic Aerosol during 2006 in Research Triangle Park, NC[J]. Aerosol and Air Quality Research, 11(2): 99–108.
|
Kleindienst T E, Conver T S, McIver C D, et al. 2004. Determination of secondary organic aerosol products from the photooxidation of toluene and their implications in ambient PM2.5[J]. Journal of Atmospheric Chemistry, 47(1): 79–100.
DOI:10.1023/B:JOCH.0000012305.94498.28
|
Kleindienst T E, Jaoui M, Lewandowski M, et al. 2007. Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location[J]. Atmospheric Environment, 41(37): 8288–8300.
DOI:10.1016/j.atmosenv.2007.06.045
|
Liang L L, Guenter E, Du Z Y, et al. 2016. Seasonal variations and source estimation of saccharides inatmospheric particulate matter in Beijing, China[J]. Chemosphere, 150: 365–377.
DOI:10.1016/j.chemosphere.2016.02.002
|
Lin Y H, Zhang H F, Pye H O T, et al. 2013. Epoxide as a precursor to secondary organic aerosol formation from isoprene photooxidation in the presence of nitrogen oxides[J]. Proceedings of the National Academy of Sciences of the Unitd States of America, 110(17): 6718–6723.
DOI:10.1073/pnas.1221150110
|
Liu Z, Wang Y H, Gu D S, et al. 2010. Evidence of reactive aromatics as a major source of peroxy acetyl nitrate over China[J]. Environmental Science & Technology, 44(18): 7017–7022.
|
Mkoma S L, Kawamura K, Fu P Q. 2013. Contributions of biomass/biofuel burning to organic aerosols and particulate matter in Tanzania, east Africa, based on analyses of ionic species, organic and elemental carbon, levoglucosan and mannosan[J]. Atmospheric Chemistry and Physics, 13(20): 10325–10338.
DOI:10.5194/acp-13-10325-2013
|
Navarro J C A, Smolander S, Struthers H, et al. 2014. Global emissions of terpenoid VOCs from terrestrial vegetation in the last millennium[J]. Journal of Geophysical Research, 119(11): 6867–6885.
|
Paulot F, Crounse J D, Kjaergaard H G, et al. 2009. Unexpected epoxide formation in the gas-phase photooxidation of isoprene[J]. Science, 325(5941): 730–733.
DOI:10.1126/science.1172910
|
Puxbaum H, Caseiro A, Sánchez-Ochoa A, et al. 2007. Levoglucosan levels at background sites in Europe for assessing the impact of biomass combustion on the European aerosol background[J]. Journal of Geophysical Research, 112(D23S05).
|
Rinne H J I, Guenther A B, Greenberg J P, et al. 2002. Isoprene and monoterpene fluxes measured above Amazonian rainforest and their dependence on light and temperature[J]. Atmospheric Environment, 36(14): 2421–2426.
DOI:10.1016/S1352-2310(01)00523-4
|
Schmidl C, Marr I L, Caseiro A, et al. 2008. Chemical characterisation of fine particle emissions from wood stove combustion of common woods growing in mid-European Alpine regions[J]. Atmospheric Environment, 42(1): 126–141.
DOI:10.1016/j.atmosenv.2007.09.028
|
Surratt J D, Chan A W H, Eddingsaas N C, et al. 2010. Reactive intermediates revealed in secondary organic aerosol formation from isoprene[J]. Proceedings of the National Academy of Sciences of the Unitd States of America, 107(15): 6640–6645.
DOI:10.1073/pnas.0911114107
|
Surratt J D, Lewandowski M, Offenberg J H, et al. 2007. Effect of acidity on secondary organic aerosol formation from isoprene[J]. Environmental Science & Technology, 41(15): 5363–5369.
|
Simoneit B R T, Schauer J J, Nolte C G, et al. 1999. Levoglucosan, a tracer for cellulose in biomass burning and atmospheric particles[J]. Atmospheric Environment, 33(2): 173–182.
DOI:10.1016/S1352-2310(98)00145-9
|
Simoneit B R T, Elias V O, Kobayashi M, et al. 2004. Sugars-dominant water-soluble organic compounds in soils and characterization as tracers in atmospheric particulate matter[J]. Environmental Science & Technology, 38(22): 5939–5949.
|
Szmigielski R, Surrat J D, Gómez González Y, et al. 2007. 3-Methyl-1, 2, 3-butanetricarboxylic acid: An atmospheric tracer for terpene secondary organic aerosol[J]. Geophysical Research Letters, 34(24): L24811.
DOI:10.1029/2007GL031338
|
Velasco E, Lamb B, Westberg H, et al. 2007. Distribution, magnitudes, reactivities, ratios and diurnal patterns of volatile organic compounds in the Valley of Mexico during the MCMA 2002 & 2003 field campaigns[J]. Atmospheric Chemistry and Physics, 7: 329–353.
DOI:10.5194/acp-7-329-2007
|
Verma S K, Kawamura K, Chen J, et al. 2015. Thirteen years of observations on biomass burning organic tracers over Chichijima Island in the western North Pacific: an outflow region of Asian aerosols[J]. Journal of Geophysical Research, 120(9): 4155–4168.
|
Volkamer R, Platt U, Wirtz K. 2001. Primary and secondary glyoxal formation from aromatics: Experimental evidence for the bicycloalkyl-radical pathway from benzene, toluene, and p-xylene[J]. Journal of Physical Chemistry, 105(33): 7865–7874.
DOI:10.1021/jp010152w
|
Wang W, Wu M H, Li L, et al. 2008. Polar organic tracers in PM2.5 aerosols from forests in eastern China[J]. Atmospheric Chemistry and Physics, 8(24): 7507–7518.
DOI:10.5194/acp-8-7507-2008
|
汪午, 王省良, 李黎, 等. 2008. 天然源二次有机气溶胶的研究进展[J]. 地球化学, 2008, 37(1): 77–86.
|
Zhang Q, Streets D G, Carmichael G R, et al. 2009. Asian emissions in 2006 for the NASA INTEX-B mission[J]. Atmospheric Chemistry and Physics, 9(14): 5131–5153.
DOI:10.5194/acp-9-5131-2009
|
Zhang Y L, Wang X M, Barletta B, et al. 2013. Source attributions of hazardous aromatic hydrocarbons in urban, suburban and rural areas in the Pearl River Delta (PRD) region[J]. Journal of Hazardous Materials, 250: 403–411.
|
Zhang Y X, Shao M, Zhang Y H, et al. 2007. Source profiles of particulate organic matters emitted from cereal straw burnings[J]. Journal of Environmental Sciences, 19(2): 167–175.
DOI:10.1016/S1001-0742(07)60027-8
|
Zheng J Y, Shao M, Che W W, et al. 2009. Speciated VOC emission inventory and spatial patterns of ozone formation potential in the Pearl River Delta, China[J]. Environmental Science & Technology, 43(22): 8580–8586.
|
Zhu C M, Kawamura K, Fukuda Y, et al. 2016. Fungal spores overwhelm biogenic organic aerosols in a midlatitudinal forest[J]. Atmospheric Chemistry & Physics, 16(11): 7497–7506.
|