 2018, Vol. 38
2018, Vol. 38



絮凝是重要的胶体化学原理之一, 也是重要的水处理方法之一.水处理中的絮凝理论包括DLVO理论、高分子吸附架桥理论、网捕卷扫理论等.众所周知, 20世纪40年代, 苏联学者Дeрягин、Ландау与荷兰学者Verwey、Overbeek分别提出了关于微粒之间的相互吸引力(能)与双电层排斥力(能)的计算方法, 据此对憎液溶胶的稳定性进行了定量处理, 被称作胶体稳定性的DLVO理论.根据DLVO理论, 微小颗粒之间的作用力是双电层排斥力与wan der Waals吸引力的加合, 表示如下:
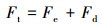
|
(1) |
式中, Ft是总相互作用力, Fe是双电层排斥力, Fd是wan der Waals吸引力, 此两种作用均与微粒间的距离有关, 所以常用相互作用势能来表示:
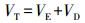
|
(2) |
式中, VT为总相互作用势能, VE为双电层排斥势能, VD为wan der Waals吸引势能.图 1即为经典DLVO理论的综合作用势能图.
 |
| 图 1 DLVO理论的综合作用势能 (VR为双电层排斥作用势能;VA是wan der Waals吸引势能;VT是综合作用势能) Fig. 1 Total interaction potential energy of DLVO theory |
图 1表示在综合作用势能曲线的远距离处存在一个第二极小值, 在近距离处存在一个第一极小值, 两个极小值中间有一个势垒M.二微粒互相靠近时, 首先到达第二极小值处, 由于第二极小值与布朗运动动能大小相近, 因而仅能发生微弱的絮凝.两微粒继续靠近可达势垒附近, 若要接近到此距离以内, 微粒的动能必须超过此势垒, 一旦超过便会越过, 就会转变为以吸引作用为主, 微粒将能继续靠近, 当接近至第一极小值处时, 表现为甚强的吸引力, 其作用远超过布朗运动动能, 微粒发生强烈聚结, 称为凝聚.
经典DLVO理论被提出后被广大研究者所接受, 成为现代絮凝科学的基础, 但之后的研究发现在具有疏水表面的微粒之间存在更强的吸引作用, 其强度超过了wan der Waals吸引力, 因此综合作用力(能)与经典DLVO理论的预计值不符, 其超过部分被认为是由表面的疏水性所引起, 称为疏水作用力.由疏水作用力引起的絮凝则被称为疏水絮凝(Pashley et al., 1981; Israelachvili et al., 1982).
疏水作用力和疏水絮凝在选矿工业领域首先得到了承认, 并得到了广泛的应用, 成为矿物加工领域20世纪最有代表性的创新成果(宋少先, 1993), 其最主要的具体做法是在浮选前的预处理中往矿物微粒的水悬浊体系中加入非极性油以强化矿粒的疏水性(Song et al., 2012, Yi et al., 2018)或加入表面活性剂在矿粒表面形成疏水性吸附层(Ucbeyiay and Ozkan, 2006), 从而增强疏水作用力, 引起疏水絮凝, 产生疏水絮体, 为后续选择性浮选创造条件.遗憾的是迄今在水处理领域, 涉及到絮凝机理时, 疏水作用力及疏水絮凝从未被提及.尽管在水环境和水处理中疏水絮凝的发生是一个事实, 如在天然水环境中悬浮及胶体颗粒吸附腐殖质、表面活性剂和碳氢化合物分子后发生的絮凝沉降, 在水处理中胶束的形成、气浮法去除杂质、隔油池除油、粗粒化聚结除油及疏水缔合絮凝剂的研究开发等均有疏水作用力及疏水絮凝机理的参与, 但研究人员和工程师并没有认识到其中疏水作用力和疏水絮凝所起的作用.这种现状可能会影响到水处理絮凝理论及技术的发展, 鉴于此本文就疏水作用力及疏水絮凝作一简要的介绍和讨论, 希望能对现今的水处理絮凝理论作一补充, 对水处理絮凝实践的发展提供有益的参考.
2 疏水絮凝和疏水作用力的发现(Discovery of hydrophobic flocculation and hydrophobic force)疏水絮凝现象可以通过一个简单的实验清楚地观察到:将纯净的石英微粒投入水中制成悬浊液, 此时石英颗粒表面是完全亲水的, 悬浊液处于稳定的分散状态.但是将这些石英微粒置于二氯二甲基硅烷蒸汽中, 就会在其表面上覆盖一层甲基硅烷, 使它表面具有很强的疏水性, 当置于水中时就会产生剧烈的团聚现象, 迅速沉向容器底部.根据颗粒表面疏水化的起因, 将疏水絮凝分为以下两种:
① 天然疏水絮凝:它是指天然疏水颗粒在水中产生团聚的现象.如细微油滴、聚四氟乙烯颗粒、石墨微粒、煤炭微粒等.这种疏水絮凝可以在不添加任何药剂的情况下发生.
② 诱导疏水絮凝:由表面活性剂分子在颗粒表面吸附导致颗粒疏水化, 进而发生疏水絮凝被称为诱导疏水絮凝.例如加十二胺于石英微粒悬浊液、加油酸钠于锡石微粒悬浊液等都可以导致其中的微粒发生团聚沉降.
疏水絮凝存在的另一个证据可以从絮凝发生时的Zeta电位看出.对多种矿物微粒悬浮体系, 在没有任何表面活性剂加入的情况下, 颗粒的聚结现象出现在Zeta电位绝对值很小处, 而在Zeta电位绝对值大的地方, 体系保持稳定的分散状态, 这恰恰是经典DLVO理论能够很好解释的现象.然而一旦油酸钠被加入矿物微粒悬浮体系, 这种现象就不复存在, 体系的聚结不是在等电点附近发生, 而是在颗粒的Zeta电位很高处出现, 显然诱导疏水絮凝是与电解质凝聚完全不同的聚结现象, 经典DLVO理论不再适用.实验证明, 矿物微粒诱导絮凝的Ea(聚结效率)~pH曲线与颗粒θ(接触角)~pH曲线有非常好的一致性, 在诱导疏水颗粒具有最大接触角的pH处, 会出现聚结效率的最大值(宋少先, 1993).
疏水絮凝的存在说明了疏水作用力的存在.Pashley和Israelachvili(Pashley et al., 1981; Israelachvili et al., 1982)测定了以阳离子表面活性剂十六烷基三甲基溴化胺(CTAB)包覆的园柱形微小云母片之间的总相互作用力, 分别以其对表面曲率半径标化后的值对表面间隔距离作曲线得图 2A.图中实线为经典DLVO理论的预测值(作了同样的标化处理), 以供比较.
 |
| 图 2 两圆柱形疏水表面之间的相互作用力与间隔距离的函数关系 (F为两表面之间的作用力;R为表面曲率半径;实线为经典DLVO理论预测值, 虚线为实验测定值) Fig. 2 Interaction force between two cylindrically curved hydrophobic surfaces as a function of distance |
图 2A显示了表面之间的作用力为表面间隔距离的函数, 其中的曲线a和曲线b分别表示表面具有不同电荷密度的情况, 插图为对未包覆表面活性剂的石英片所测得的作用力.可以看出, 在间隔距离较远处测定值与DLVO预测值能很好吻合, 随着距离的减小, 测定值逐渐偏离了DLVO预测值, 最后大大超过了预测值, 显示了更强的吸引作用.插图中对未包覆表面活性剂的石英片所测得的作用力与DLVO理论预测值有极好的符合性. Israelachvili和Pashley认为, 在包覆表面活性剂的石英片之间所测得的作用力超过wan der Waals吸引力的额外部分即为疏水作用力, 从DLVO理论值减去实测值就得到疏水作用力Fh, 这种作用力作用范围比普通共价键的作用要长, 也比经典van der Waals作用力长, 约在1~10 nm之间, 甚至某些情况下在大于100 nm的间距处都可测得(Hammer et al., 2010), 因而被称为长程作用力.以Fh对表面曲率半径作标化计算得Fh/R, 并对间隔距离作对数线(图 2B), 可以看出, 疏水作用按指数规律下降至10 nm的间距处, 由此得到以指数函数规律随距离衰减的疏水作用力的经验式:
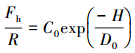
|
(3) |
式中, Fh是园柱状云母片之间的疏水作用力, R是云母片的曲率半径, H是云母片之间的最短距离, C0及D0为拟合参数, C0数值的大小决定着疏水作用力的强弱, D0具有长度单位, 其大小决定疏水作用力作用距离的范围, 被称为衰减长度.式(3)表示疏水作用力随微粒之间的最短距离按指数函数规律逐渐衰减.Claesson等(1988)及Yoon等(1996)发现当使用不溶性双碳氢链表面活性剂包覆小云母片时, 产生的疏水作用力更强, 衰减距离范围更长, 此时采用以下双指数函数能够更好地符合实验数据:
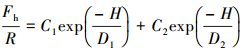
|
(4) |
式中, C1、C2、D1、D2与式(3)中的C0、D0相同, 但数值不同.
自30多年前疏水作用力被发现, 时至今日仍不断有相关研究报道(Parker and Claesson, 1994; Stock et al., 2015; Yuhei et al., 2015; Ducker et al., 2016; Yi et al., 2018), 均证明了疏水作用力是客观存在, 其中也有一些研究者主张采用幂函数的经验公式(Rabinovich and Yoon, 1994; Pazhianur and Yoon, 2003):
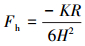
|
(5) |
式中, K是单一拟合参数.该式表示疏水作用力随微粒之间的最短距离按幂函数规律衰减, 与wan der Waals吸引力计算式(Chang, 2016)有相同的形式, 研究者可以直接将K与wan der Waals式中的Hamaker常数的数值进行比较.以十八烷基三氯硅烷包覆硅片进行的试验得到K值在(0.11~3.5)×10-16 J之间, 比硅片在水中的Hamaker常数(~10-20 J)大数个数量级, 由此同样可说明疏水作用力的存在.
基于疏水作用力的发现, 传统的DLVO理论应修正为扩展的DLVO理论, 其数学表达式为:
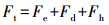
|
(6) |
式中, Ft为微粒之间的总相互作用力, 它等于静电作用力Fe、van der Waals作用力Fd及疏水作用力Fh三者之和.
3 疏水作用力产生的机理(Mechanism of hydrophobic force origination)尽管疏水作用力在实验上得到了证实, 但迄今对它产生的机理还不甚清楚, 尚存在一些争论.从热力学上讲可以解释如下(宋少先, 1993;Ducker et al., 2016):水分子间氢键的破坏是导致疏水作用力产生的原因.当疏水颗粒或非极性分子进入水中时, 水分子间的氢键结构将遭到部分破坏而断裂, 那些与疏水颗粒表面或非极性分子相邻的水分子不能与之形成氢键, 因而能量会升高, 按照热力学定律, 能量高的体系是不稳定的, 必然会向能量低的状态转化, 因此疏水颗粒或非极性分子周围的水分子总是企图将其周围的疏水颗粒或非极性分子排斥开, 或自身从疏水表面之间的范围流出进入本体相, 以恢复氢键缔合的结构, 由此形成所谓“疏水作用力”, 迫使疏水颗粒或非极性分子相互聚结或逃离水体内部而在气液界面集结, 以减小疏水颗粒或非极性分子与水的接触面积, 使体系的自由能降低.由于水分子在水中以氢键缔合方式形成网状结构, 所以疏水颗粒在疏水界面上发生的扰动会从界面向水中传播, 传播范围大于数个水分子直径, 因此疏水作用力被称为长程作用力(Ducker et al., 2016).目前对疏水作用力产生的微观机理的解释主要有空化作用机理和偶极耦合机理, 分别介绍如下.
3.1 空化作用Bérard等(1993)对限制于刚性平滑疏水表面之间的液体采用Monte Carlo方法作了数值模拟.Monte Carlo方法适合于寻找两相平衡共存点, 找出临界亚稳态点, 可以用来研究超过平衡共存点的亚稳态液相.模拟结果表明, 疏水作用力是由疏水表面的微小间隔距离所导致的相变所引起:一种液体虽然在处于主流体中或被限制于间隔距离较大的两刚性平滑表面之间时为液相, 但当表面相互接近到间隔距离极小时, 则会变为亚稳态, 此亚稳态水发生毛细蒸发成为气体(恰与毛细凝结现象相反), 形成空穴, 即空化作用, 由此造成密度减小和Laplace压力差, 导致两表面之间的相互吸引力, 其强度数倍于wan der Waals力, 属长程作用力, 并在两表面接近至亚稳态间距时迅速增大.另据平均场分析, 由于疏水表面的导入, 极性液体被惰性表面代替, 当表面接近至一定程度时, 表面之间的液体的化学势开始降低, 相变随之发生, 由化学势开始降低点可以确定临界亚稳态间距.Bérard等(1993)由平均场分析和Monte Carlo模拟得到净压力的数学表示式如下:
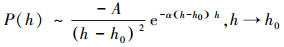
|
(7) |
式中, A、α为常数, h为两疏水表面间距, h0为达到临界压稳态时的间距.模拟结果如图 3和图 4所示.
 |
| 图 3 疏水表面之间与主流体平衡的流体的Monte Carlo平均密度 (图中插图是对图下方气体行为的放大; σ分子直径, 纵坐标物理量是表示密度大小的一种量) Fig. 3 Monte Carlo average density for the fluid between hard walls and in equilibrium with bulk |
 |
| 图 4 刚性平滑表面之间的净压力 (P是压力, β和σ均为常数, 纵坐标所示物理量是表示压力大小的一种量,实线表示van der Waals吸引作用力, 虚线是实验数据拟合平均场结果) Fig. 4 The net pressure between hard walls |
图 3表示密度变化, 其表达式为

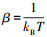
图 4表示压力变化, 其中图 4a为第一状态点(βAσ=0.15, ασ2=0.02, h0=3.0σ)的情况, 图 4b为第二状态点(βAσ=0.13, ασ2=0.02, h0=3.0σ)的情况.从图 4看, 两表面之间的净压力为负值, 表明该力是吸引力, 并随表面间隔距离的增大而减弱.所谓净压力等于总压力减去主流体的压力.在气液两相共存的间隔距离处, 当液体蒸发为气体时, 表现为很强的吸引作用.图中虚线表示液体的实验数据拟合式(7)的结果, 实线表示van der Waals作用力的计算值, 可以看出, DLVO理论的van der Waals作用力的计算值大大低估了亚稳态液体所导致的吸引力.
综上所述, 空化作用理论认为, 当液体中惰性表面相互接近时, 其间隔距离变小会导致液体变为亚稳态, 亚稳态液体通过毛细蒸发形成气穴, 即空化作用, 由此产生密度差和Laplace压力差, 从而导致使疏水颗粒聚结的疏水作用力, 此疏水作用力为长程作用力, 其值随着空化距离的临近迅速增大.
3.2 偶极相互作用Yoon等(1996)认为, 表面活性剂分子在浓度低于临界胶束浓度CMC的情况下可以形成有序排列的半胶束, 它们以—CH3朝向水中的方向垂直吸附在微粒表面, 形成单分子层的偶极膜块, 而邻近水分子由于失去了氢键会以单一定向平行排列方式吸附于此膜块上, 与之共同形成大偶极.这样形成的大偶极会与相邻颗粒上同样生成的大偶极相互吸引而产生疏水作用力.根据此理论, 疏水作用力本质上应属于静电力.
Pazhianur等(2003)根据上述理论针对硅烷化的硅片与硅烷化的玻璃球之间的疏水作用力提出了如下计算式:
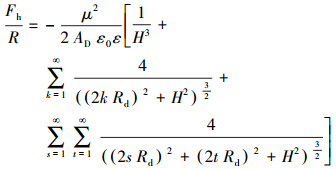
|
(8) |
式中, 
此后, Pazhianur等(2003)以十八烷基三氯硅烷对硅片及玻璃小球作了表面硅烷化处理, 使硅片的前进接触角分别为0°、75°、83°和92°, 并以原子力显微镜(AFM)测定了这些硅烷化硅片与硅烷化玻璃球之间在不同距离的吸引力, 将所得吸引力数据以玻璃球半径标化后按扩展的DLVO理论式(6)拟合, 此处式(6)中Fh按公式(8)计算, 得图 5a.
 |
| 图 5 硅烷化硅片与硅烷化玻璃球之间相互作用的F/R-H曲线 (a.接触角较小, 仅考虑了偶极耦合作用, 未考虑空化作用, b.接触角较大, 考虑了偶极耦合及空化作用; θa是前进接触角; 虚线是将数据拟合为DLVO理论的曲线, 实线是将数据拟合为扩展的DLVO理论的曲线) Fig. 5 The F/R-H curves obtained for the interaction of silanated glass spheres and silica plates |
在图 5a中, 由于硅烷化处理不影响表面热力学电位, 上述4种前进接触角情况下的DLVO理论拟合线相同, 均以同一虚线代表.3条实线则分别代表除0°外的3种前进接触角的情况下扩展的DLVO理论拟合曲线, 其中Fh按公式(8)计算.结果表明, 每一种接触角情况下的测定值都与经典DLVO计算值不同, 其中, 在前进接触角为0°时表现出额外的近距排斥力, 作者认为这是由表面水化层所导致, 而其它3种前进接触角的情况下, 均表示可以很好地拟合为扩展的DLVO理论, 比经典DLVO理论预见值有更强的吸引力, 此额外的吸引力即未被经典DLVO理论所考虑的疏水作用力.
在研究了前进接触角小于90°的情况后, Pazhianur等(2003)再次使用十八烷基三氯硅烷对玻璃小球和硅片作了表面硅烷化处理, 得到了前进接触角大于90°的小球和小片, 对前进接触角分别为109°、105°和100°的情况, 以原子力显微镜(AFM)测定了它们之间在不同间隔距离处的吸引力, 但得到了与接触角小于90°的情况不同结果:在较远的间距处, 数据依然能很好地拟合为公式(6), 但在较近处却产生了较大的偏差, 实验测得的吸引力显著大于公式(6)的预见值, 表明不但不符合经典DLVO理论, 也不符合扩展的DLVO理论.
对于较近间隔距离处实验值与扩展的DLVO理论值之间的较大偏差, Pazhianur等(2003)认为, 在近距离处必有另一种作用力存在, 这种作用力在扩展的DLVO公式(6)和式(8)中没有被考虑, 而引起这种作用力的原因是在疏水表面附近形成的气穴, 即发生了空化作用.基于此, Pazhianur和Yoon在公式(8)中加上了气穴贡献项, 如式(9)所示:
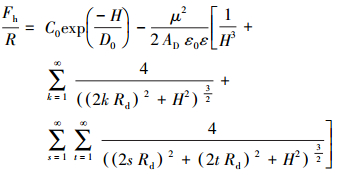
|
(9) |
式中, 第一项(即指数项)表示空化作用的贡献, 第二项表示表面膜块大偶极的贡献.根据此式, 将原子力显微镜(AFM)测定所得硅烷化的硅片与硅烷化的玻璃球之间在不同距离的吸引力数据拟合为式(6), 其中Fh按公式(9)计算, 得图 5b.
可以看出, 扩展的DLVO理论在加上了空化作用的贡献项后能够在全程衰减范围内非常好地与实验数据符合, 所以对于较大的接触角, 即疏水性更强的表面, 偶极相互作用与空化作用均应给于考虑.
空化机理和偶极耦合机理的研究进一步说明疏水作用是存在的, 它既不是化学键力, 也不是分子间力.目前关于疏水作用产生的微观机理尚不完善, 还有不同的看法(Mishchuk, 2011), 但偶极相互作用和空化作用应该是目前的主流观点.无论机理如何, 众多研究者的工作说明疏水作用力的存在是不可否认的事实.
4 水处理中的疏水絮凝(Hydrophobic flocculation in water treatment)尽管疏水絮凝是一个普遍存在的现象, 但长期以来人们没有认识到它, 而是把它与电解质凝聚及高分子絮凝混为一谈, 严重影响了疏水絮凝在科学技术领域中的应用, 特别是在水处理领域, 一些科研工作者没有能认识到疏水絮凝与电解质凝聚的本质区别, 耗费精力去寻找电解质对疏水絮凝的影响, 或牵强附会地将它解释为高分子架桥作用, 因而一无所获.20世纪80年代以后, 人们对疏水颗粒在水中产生团聚的现象及其原理的认识进入了一个新的阶段, 使疏水絮凝在许多领域得到了普遍的重视和越来越多的应用, 例如在选矿界开发了以诱导疏水絮凝原理为基础的微细矿粒分选工艺, 取得了显著的效益.与电解质凝聚和高分子絮凝相比, 疏水絮凝有诸多优点, 例如产生的絮体结构比较密实, 空隙较小, 强度较高, 絮凝过程具有可逆性, 被外力破坏的絮体可在适当的水力条件下重新聚结成团, 絮体的水分含量较低, 有利于污泥脱水作业等, 都是水处理絮凝单元力求达到的效果.事实上在水处理的许多已有方法中已经涉及到疏水絮凝原理, 需要水处理科研工作者认识它, 面对它, 进一步利用并扩大其效能.
4.1 气浮法中的疏水絮凝在工业废水的处理中, 经常遇到一类废水, 其中所含的悬浮颗粒或胶体污染物的直径很小, 密度接近或小于水, 如果用沉降法去除这些悬浮物, 往往效果很差.对于这些种类的废水, 气浮法是一种有效的固液或液液分离手段(高廷耀等, 2015;Chang, 2016), 被广泛地用来去除水中的某些污染物, 如天然水中的藻和植物残体、印染废水中的染料颗粒、造纸化纤工业废水中的短纤维、炼油化工废水中的细微油滴、生活污水中的油脂及蛋白质等、污水处理产生的活性污泥、工业废水中的重金属离子、阴离子、表面活性剂、橡胶、树脂等(Rubio et al., 2007).对这些污染物的气浮工艺实际是絮凝和浮上的组合, 在絮凝单元要加入某种无机或有机高分子絮凝剂, 在浮上单元以某种方法(如空气压缩机、真空泵)在水中产生细小分散的气泡.加入的无机或有机高分子絮凝剂使污染物颗粒聚结形成絮体(Karhu et al., 2014; Saththasivam et al., 2016), 由于高分子絮凝剂一般具有疏水碳氢链, 所以疏水絮凝对絮体的形成有一定的贡献, 形成的絮体又具有一定的疏水性, 疏水性絮体易黏附于微气泡上, 随之上浮至水面而被分离.这里有机高分子絮凝剂具有疏水链段是有利条件, 但有机高分子絮凝剂常常同时具有亲水基团(如阴离子型和阳离子型聚丙烯酰胺), 所以絮体的疏水性相对较弱.在矿物分选工艺中一般是加入表面活性剂, 表面活性剂具有碳氢链较短和疏水性较强的特点, 其分子以其极性基吸附于污染物颗粒之上, 以其疏水碳氢链伸向水中, 在疏水链段的疏水作用力下, 矿物微粒发生诱导疏水絮凝成为较大的絮体, 有利于进一步吸附和截留气泡, 碳氢链的疏水性越强, 就越容易被气泡黏附, 这实际上是固气间的疏水作用力引起的一种疏水絮凝, Moreno-Atanasio (2013)考察了实际数据后, 提出了3种模式来描述气固间的疏水作用力随间隔距离的变化规律, 若以d表示微粒与气泡的间距, 第一种模式中疏水作用力随间距的增大按1/d规律衰减, 第二种模式按1/d2规律衰减, 第三种按指数规律exp(-d/λ)衰减, 其中λ为衰减长度.
4.2 粗粒化法及隔油池处理技术中含油废水的疏水絮凝含油废水对环境有着各种不同的危害.含油废水中的油有4种形态, 即溶解油、乳化油、分散油及浮油.含油废水的预处理方法常采用粗粒化法和隔油池法(张文林等, 2005).粗粒化法和隔油池法处理的对象主要是水中的分散油.
在粗粒化法除油过程中, 分散油珠在聚结器内发生聚结, 然后在油水分离器内被分离.聚结过程中废水中的油珠颗粒发生凝并, 形成较大颗粒, 从而提高油水分离的效率, 其机理分为碰撞聚结和润湿聚结.碰撞聚结指油珠在液相中相互碰撞合并成为大的油珠, 润湿聚结是液相中的油珠与疏水亲油介质黏附后发生聚结, 或与已黏附在介质表面的油珠或油膜发生碰撞后产生凝并, 无论是碰撞聚结还是润湿聚结, 其驱动力都是疏水作用力, 实际都应属于疏水絮凝的范畴.
在隔油池中, 废水以较低的流速在池中作水平流动, 密度小于水的油珠杂质将逐渐上升至水面, 在流动过程中逐渐凝并为浮油, 在到达隔油池末端后进入集油管而被分离.在隔油池中分散油的凝并也是在疏水作用力的驱动下发生的, 应属于疏水絮凝的范畴.
4.3 疏水缔合型絮凝剂的研究开发及其疏水絮凝机理自21世纪初起, 高分子疏水基团的缔合作用引起了众多研究者的兴趣.胡红旗等(2003)综述了在水溶性高分子上引入疏水基团的研究工作, 指出所得高分子的疏水缔合作用是疏水基团在疏水作用和亲酯作用下发生的碳氢链之间的簇集, 疏水缔合作用会引起高分子溶液流变性质的改变.在此研究的启发下, 国内水处理工作者开始研究开发疏水缔合絮凝剂.
陈鸿等(2003)以疏水性单体丙烯酸丁酯与丙烯酰胺、二甲基二烯丙基氯化铵等合成了疏水改性阳离子型高分子絮凝剂, 并用它处理了硅藻土悬浊液, 研究者认为分子中的疏水基团增强了高分子絮凝剂的吸附架桥作用.
钟传蓉等(2007)以疏水缔合作用强且耐酸碱的丁基苯乙烯(BS)为疏水单体, 与丙烯酰胺(AM)、二甲基二烯丙基氯化铵(DMDAAC)合成了AM-BS-DMDAAC共聚物PBAD, 然后以硅藻土悬浊液为絮凝对象进行了除浊实验, 根据实验结果认为含有疏水基团的阳离子型聚合物能在水溶液中发生缔合形成网络结构, 从而提高了絮凝剂的吸附架桥能力和对水体中有机物的去除能力.
李刚辉等(2008)以含有多个—CF3基团的甲基丙烯酸十二氟庚酯为疏水改性单体对聚丙烯酰胺阳离子絮凝剂改性, 制备了疏水性高分子絮凝剂, 并用于造纸白水的絮凝处理.实验表明, 絮凝剂分子的疏水侧链可发生分子间缔合和分子内缔合.作者认为这可以增强吸附架桥作用.
鲁智勇等(2008)以疏水单体CX-甲基丙烯酰氧乙基三甲基氯化铵(CXDMC)及丙烯酰胺、甲基丙烯氧乙基三甲基氯化铵(DMC)为原料, 制备了疏水缔合阳离子絮凝剂P(AM-DMC-CXDMC), 通过对硅藻土悬浊液的絮凝实验证明其有较好的絮凝效能, 作者将其原因归于疏水分子链的缔合所导致的吸附架桥作用增强.
朱庆胜等(2009)以疏水单体全氟辛基乙基丙烯酸酯(FM)及2-丙烯酰胺基-2-甲基丙磺酸(AMPS)、丙烯酰胺(AM)和甲基丙烯酰氧乙基三甲基氯化铵(DMC)等为原料, 通过自由基胶束共聚合成了P(FM-AMPS-AM-DMC)四元共聚物, 即氟碳型两性聚丙烯酰胺(FPAM)絮凝剂.实验结果表明, FPAM水溶液中存在强烈的分子间缔合作用, 用该絮凝剂处理洗煤废水时, 电中和与吸附架桥为主要作用机理.
郭晓丹等(2015)通过在糊化淀粉上引入疏水单体甲基丙烯酰氧乙基二甲基十六烷基溴化铵(CDMN)及丙烯酰胺(AM)、功能性阳离子单体二甲基二烯丙基氯化铵(DMDAAC)和纳米SiO2制备了疏水缔合阳离子改性淀粉-纳米SiO2复合絮凝剂(CSSADD), 并针对高岭土悬浊液与其它絮凝剂作了絮凝性能比较, 实验证明在絮凝剂分子上引入疏水基团增强了其絮凝性能, 作者认为机理是疏水基团可增强絮凝剂的吸附架桥作用.
Liao等(2014)考查了近年来在疏水改性聚丙烯酰胺的合成及水处理应用方面的进展, 认为将疏水基团引入分子结构可以使聚合物分子链之间的作用加强, 使聚合物与有机物之间的作用加强, 使絮体的的亲水性减弱, 因而可以得到较好的絮凝效果.
刘孔怡等(2017)以环氧氯丙烷、乙二胺和十六烷基二甲基叔胺为原料, 合成了以聚环氧氯丙烷-乙二胺为亲水主链, 长链叔胺为疏水侧链的聚环氧氯丙烷胺类疏水缔合型絮凝剂, 并用它处理了高岭土悬浊液, 作者认为作用机理是:带有正电荷的亲水链与水中带负电荷的杂质颗粒发生电中和, 而聚合物分子在疏水缔合作用下相互缠结, 形成大块絮团, 快速沉降而使水澄清.
总结国内疏水缔合高分子絮凝剂的研究情况可以发现, 大多数国内研究者都认为疏水缔合高分子絮凝剂的作用机理是高分子中引入的疏水基团增强了其吸附架桥作用.笔者认为, 决定吸附架桥作用的因素有二, 一是高分子上有具有吸附性能的极性基团, 二是高分子链的长度及伸展度, 而不是疏水基团的存在, 理由是疏水基团不能在亲水性高岭土等微粒上发生吸附, 也不喜伸入水中而在分子内缔合, 不利于其伸展, 因而不利于架桥作用.由于以上高分子絮凝剂的链长对架桥作用已足够, 所以分子间缔合对增强架桥作用的影响是有限的.研究者应该认识到疏水缔合高分子絮凝剂的絮凝增强机理应该有疏水作用力引起的疏水絮凝的贡献.上述研究中只有刘孔怡认为疏水缔合高分子絮凝剂的作用机理是聚合物分子在疏水缔合作用下相互缠结, 形成大块絮团, 快速沉降而使水澄清, 可以说刘孔怡的观点是比较接近疏水絮凝机理的, 但其未提及疏水作用力及疏水絮凝机理.
5 结论(Conclusions)精确的实验测量证明疏水作用力是客观存在, 在疏水作用力的作用下疏水性颗粒会发生疏水絮凝.考虑疏水作用力和疏水絮凝, 经典的DLVO理论应补充为扩展的DLVO理论.疏水作用力及疏水絮凝的热力学解释是, 当具有疏水表面的微粒被引入水中时, 水分子的氢键缔合结构会遭到破坏, 导致临近界面的水分子能量升高.为降低体系能量, 疏水界面之间的水分子会排斥疏水颗粒, 或采取自身流出界面之间范围的方式, 迫使疏水颗粒聚结, 发生絮凝.目前对疏水絮凝产生的微观机理的认识主要有空化作用和偶极耦合作用, 但理论尚不完善, 还有不同看法, 尚须进一步探讨.
6 期望(Expectation)事实上在水处理领域的许多技术中早已存在或涉及到疏水作用力和疏水絮凝, 目前特别需要水处理工作者以疏水絮凝的理论去认识它们, 针对水污染控制的任务深入开展疏水絮凝的基础理论和应用技术的研究, 例如覆盖率及疏水率对疏水作用的影响及其机理、疏水絮凝适用的水质种类及水质条件、疏水絮凝的实施方法及操作条件、疏水絮凝与高分子絮凝、电解质凝聚等传统方法的比较和客观评价, 开发性能更优的疏水缔合性絮凝剂及表面活性剂等, 从而提升疏水絮凝效果, 尽快实现疏水絮凝在水处理实践中的应用.
Bérard D R, Attard P, Patcy G N. 1993. Cavitation of a Lennard-Jones fluid between hard walls, and the possible relevance to the attraction measured between hydrophobic surfaces[J]. Journal of Chemical Physics, 98(9): 7236–7244.
|
陈鸿, 张熙, 梁兵. 2003. 疏水改性阳离子型高分子絮凝剂P(AM-DMDAAC-BA)的合成与性能研究[J]. 高分子材料科学与工程, 2003, 19(2): 97–100.
DOI:10.3321/j.issn:1000-7555.2003.02.024 |
Chang Q. 2016. Colloid and Chemistry for Water Quality Control[M]. London: Elsevier. 111; 239-240
|
Claesson P M, Christenson H K. 1988. Very long range attractive forces between uncharged hydrocarbon and fluorocarbon surfaces in water[J]. Journal of Chemical Physics, 92(6): 1650–1655.
|
Ducker W A, Mastropietro D. 2016. Forces between extended hydrophobic solids:Is there a long-range hydrophobic force?[J]. Current Opinion in Colloid & Interface Science, 22: 51–58.
|
高廷耀, 顾国维, 周琪. 2015. 水污染控制工程(第4版)[M]. 北京: 高等教育出版社: 66–71.
|
郭晓丹, 诸林, 焦文超. 2015. 疏水缔合阳离子改性淀粉-纳米SiO2絮凝剂CSSADD的制备和性能测试[J]. 精细化工, 2015, 32(12): 1402–1407.
|
胡红旗, 陈鸣才, 程镕时. 2003. 疏水缔合作用研究[J]. 广州化学, 2003, 28(1): 26–33.
DOI:10.3969/j.issn.1009-220X.2003.01.007 |
Hammer M U, Anderson T H, Chaimovich A, et al. 2010. The search for the hydrophobic force law[J]. Faraday Discussions, 146(1): 299–308.
|
Isrelachvili J, Pashley R. 1982. The hydrophobic interaction is long range, decaying exponentially with distance[J]. Nature, 300(25): 341–342.
|
Karhu M, Leiviskä T, Tanskanen J. 2014. Enhanced DAF in breaking up oil-in-water emulsions[J]. Separation & Purification Technology, 122(3): 231–241.
|
李刚辉, 沈一丁, 李培枝, 等. 2008. 氟碳改性阳离子PAM的溶液性能及应用[J]. 高分子材料科学与工程, 2008, 24(8): 116–119.
DOI:10.3321/j.issn:1000-7555.2008.08.030 |
鲁智勇, 安利, 操卫平, 等. 2008. 疏水缔合阳离子型高分子絮凝剂的合成与性能研究[J]. 环境科学与技术, 2008, 31(10): 95–97.
DOI:10.3969/j.issn.1003-6504.2008.10.025 |
Liao Y, Zheng H, Dai L, et al. 2014. Hydrophobically modified polyacryamide synthesis and application in watertreatment[J]. Asian Journal of Chemistry, 26(18): 5923–5927.
DOI:10.14233/ajchem
|
刘孔怡, 刘江辉, 卢继雷, 等. 2017. 疏水缔合型絮凝剂的制备及性能研究[J]. 化学研究与应用, 2017, 29(10): 1483–1486.
DOI:10.3969/j.issn.1004-1656.2017.10.005 |
Moreno-Atanasio R. 2013. Influence of hydrophobic force model on capture of particles by bubbles:A computational study using discrete element method[J]. Advanced Powder Technology, 24: 786–795.
DOI:10.1016/j.apt.2013.05.001
|
Mishchuk N A. 2011. The model of hydrophobic attraction in framework of classical of DLVO forces[J]. Advances in Colloid and Interface Science, 168(1/2): 149–165.
|
Pashley R M, Israelachvili J N. 1981. A comparison of surface forces and interfacial properties of mica in purified surfactant solutions[J]. Colloids and Surfaces, 2(2): 169–187.
DOI:10.1016/0166-6622(81)80006-6
|
Pazhianur R, Yoon R H. 2003. Model for the origin of hydrophobic force[J]. Minerals and Metallurgical Processing, 20(4): 178–184.
|
Parker J L, Claesson P M, Attard P. 1994. Bubbles, cavities, and the long-ranged attraction between hydrophobic surfaces[J]. Journal of Physical Chemistry, 98(34): 8468–8480.
DOI:10.1021/j100085a029
|
Rubio J, Carissimi E, Rosa J J. 2007. Flotation in water and wastewater treatment and reuse:recent trends in Brazil[J]. International Journal of Environment & Pollution, 30(2): 197–212.
|
Saththasivam J, Loganathan K, Sarp S. 2016. An overview of oil-water separation using gas flotation systems[J]. Chemosphere, 144(1): 671–680.
|
Stock P, Utzig T, Valtiner M. 2015. Direct and quantitative AFM measurements of the concentration and temperature dependence of the hydrophobic force law at nanoscopic contacts[J]. Journal of Colloid and Interface Science, 446(11): 244–251.
|
宋少先. 1993. 疏水絮凝理论与分选工艺[M]. 北京: 煤炭工业出版社: 5–8.
|
Song S, Zhang X, Yang B, et al. 2012. Flotation of molybdenite fines as hydrophobic agglomerates[J]. Separation & Purification Technology, 98(3): 451–455.
|
Ucbeyiay H, Ozkan A. 2006. Hydrophobic flocculation characteristics of calcite and effects of some inorganic dispersants[J]. Indian Journal of Chemical Technology, 13: 448–454.
|
Yoon R H, Ravishankar S A. 1996. Long-range hydrophobic forces between mica surfaces in dodecylammonium chloride solutions in the presence of dodecanol[J]. Journal of Colloid & Interface Science, 179(2): 391–402.
|
Yuhei S, Hiroyuki I, Koreyoshi I, et al. 2015. Effect of surface hydrophobicity on short-range hydrophobic attraction between silanated silica surfaces[J]. Advanced Powder Technology, 26: 1729–1733.
DOI:10.1016/j.apt.2015.10.017
|
Yi H, Zhao Y, Rao F, et al. 2018. Hydrophobic agglomeration of talc fines aqueous Suspensions[J]. Colloids & Surfaces A Physicochemical & Engineering Aspects, 538: 327–332.
|
张文林, 李春利, 侯凯湖. 2005. 含油废水处理技术研究进展[J]. 化工进展, 2005, 24(11): 1239–1243.
DOI:10.3321/j.issn:1000-6613.2005.11.008 |
钟传蓉, 何文琼, 赖立. 2007. 罗平亚, 疏水改性阳离子型丙烯酰胺共聚物的微结构与絮凝性能[J]. 化工学报, 2007, 58(8): 2138–2143.
DOI:10.3321/j.issn:0438-1157.2007.08.042 |
朱胜庆, 李小瑞, 李培枝. 2009. 氟碳型两性聚丙烯酰胺絮凝剂的制备及性能[J]. 石油化工, 2009, 38(11): 1219–1224.
DOI:10.3321/j.issn:1000-8144.2009.11.015 |