 2018, Vol. 38
2018, Vol. 38



2. 中国科学院大学, 北京 100049
2. University of Chinese Academy of Sciences, Beijing 100049
微生物气溶胶在自然环境中普遍存在(Després et al., 2012;房文艳等, 2015;方治国等, 2005).通常, 微生物气溶胶含有细菌和真菌等微生物粒子, 按其种类被分为细菌气溶胶和真菌气溶胶.细菌气溶胶粒径范围为0.5~100 μm, 芽孢杆菌(Bacillus)、假单胞菌(Pseudomonas)、鞘氨醇单胞菌属(Sphingomonas)、葡萄球菌(Staphylococcus)等为常见的细菌种类(于玺华, 2002).有研究证实, 污水处理过程有微生物气溶胶的产生和逸散(刘建伟等, 2013;Han et al., 2013;Brandi et al., 2000;Li et al., 2013).由于污水及其处理设施中的微生物以细菌为主, 因此, 污水处理厂微生物气溶胶的研究对细菌群落结构更为关注, 特别是污水中存在病原菌和潜在致病细菌.因此, 要解析污水处理厂微生物气溶胶中细菌群落多样性, 选择适宜的采样和分析方法是关键.
根据采样器的原理, 微生物气溶胶采样方法分为自然沉降法、射流撞击式采样法、离心式采样法、过滤式采样法和静电沉降式采样法(于玺华, 2002;李涛, 2003).其中, 固体多级撞击式的Andersen采样器具有操作简便、采样效率高、微生物存活率高等优点, 通常作为可培养微生物气溶胶采集的标准方法被广泛应用(Xu et al., 2013).此外, 过滤式采样法具有抽气设备流量大、收集效率高等优点, 其中, 大流量的总悬浮颗粒物采样器常应用于基于非培养法的微生物气溶胶分子生物学研究(Cao et al., 2014).在分析方法方面, 传统的培养法在研究微生物气溶胶的微生物多样性方面存在局限性, 只能检测可培养微生物, 其数量不到微生物总数的10%(Dong et al., 2016;Urbano et al., 2011).近年来, 随着现代分子生物学技术的快速发展, 空气微生物群落的解析方法也从培养法向非培养法发展(方治国等, 2016).16S rDNA克隆文库技术(Urbano et al., 2011;Han et al., 2012)、聚合酶链式反应-变性凝胶梯度电泳(Polymerase chain reaction- denatured gradient gel electrophoresis, PCR-DGGE)(Xu et al., 2013)、基因芯片技术(Peccia et al., 2006;方治国等, 2016)、末端限制性酶切片段长度多态性分析(Terminal restriction fragment length polymorphism, T-RFLP)(方治国等, 2016)、荧光原位杂交技术(Fluorescence in situ hybridization, FISH)(Lange et al., 1997)、定量PCR技术(Wery et al., 2008;Yamamoto et al., 2014)和空气微生物宏基因组学(Cao et al., 2014;Yamamoto et al., 2014;Dannemiller et al., 2014)等技术在环境细菌气溶胶和真菌气溶胶的群落组成鉴定、丰度的确定、对特异性的致病细菌或病毒的定量检测中已有应用.此外, 指纹图谱技术、核酸杂交技术及高通量测序技术可以直接在分子水平上全面分析复杂环境微生物群落结构及多样性(Cao et al., 2014;Kumaraswamy et al., 2014).但如何解析污水处理厂细菌气溶胶目前尚没有标准方法.
本研究以某城市污水处理厂为研究对象, 以收集气溶胶中全部细菌效率最高的总悬浮颗粒物采样器进行样品收集, 并利用高通量测序技术对样品中全部细菌的多样性与群落结构进行分析.作为对照, 同步采用收集气溶胶中可培养细菌效率最高、应用最广泛的Andersen六级采样器采样, 用克隆文库技术进行分析, 以对比高通量测序与传统的培养法对同一采样点细菌气溶胶解析的异同, 为确定适宜污水处理厂细菌气溶胶的分析方法提供科学依据.
2 材料与方法(Materials and methods) 2.1 采样点细菌气溶胶样品采自某城市污水处理厂, 该污水处理厂采用SBR工艺处理生活污水, 规模为5.0×104 m3·d-1.细菌气溶胶采样点沿污水处理工艺布置, 包括细格栅、粗格栅、沉砂池、SBR池和污泥脱水间.另外, 选取污水处理厂外的上风向200 m和下风向200 m处作为对照点.采样高度设置距地面1.5 m处(人呼吸高度).
2.2 采样方法总悬浮颗粒物采样器:采用智能中流量便携式总悬浮颗粒物采样器(TH-150C, 武汉天虹仪表有限责任公司), 采样膜为直径90 mm的石英滤膜(PALL, NY, U.S.), 其对颗粒物的截留率为99.9%, 采样流量为100 L·min-1, 采样时间不低于4 h.不同采样点的细菌气溶胶样品同步采集.采样完毕, 用无菌镊子取出采样膜, 保存在无菌锡箔纸内, 尽快转运到实验室立即处理, 或者冻存于-80 ℃冰箱内.
Andersen采样器:采用Andersen六级固体撞击式采样器(228-9530 K, SKC Gulf Coast Inc., USA), 由六级带有微小喷孔的铝合金圆盘撞击器组成, 每级撞击器有400个孔, 孔的直径逐渐缩小, 模拟人体呼吸道的解剖结构和空气动力学特征, 采用惯性撞击原理将空气中悬浮的微生物粒子按照粒径大小分等级地收集到采样载体表面, 圆盘下方放盛有采样介质的培养皿, 本研究选取直径为90 mm的营养琼脂平板(02-275, 北京奥博兴生物技术有限责任公司)作为细菌气溶胶分级截留介质, 设置抽气速率为28.3 L·min-1, 样品采集时间为3 min, 采集过程中设置3个平行.
2.3 分析方法 2.3.1 DNA提取总悬浮颗粒物中全细菌DNA提取:在无菌条件下, 将总悬浮颗粒物采样器收集在石英滤膜上的颗粒物样品剪碎, 用40 mL无菌1×PBS缓冲液洗脱颗粒物, 置于4 ℃低温低速离心机中, 200 g离心2 h.通过真空过滤装置, 将上述离心后的缓冲液浓缩颗粒物至聚醚砜滤膜(PES)上, 剪碎此滤膜立即加入到MO-BIO PowerSoil DNA试剂盒(Carlsbad, CA, U.S.)中的PowerBead Tube中, 按参考文献报道的优化提取方法提取DNA(Jiang et al., 2015).
可培养细菌DNA提取:通过Andersen采样器收集细菌培养基上的颗粒物, 30 ℃恒温培养48 h后, 使用1×PBS缓冲液收集所有细菌培养物, 采用核酸自动提取仪(TanBead, 台湾)对细菌总DNA进行提取.
2.3.2 总悬浮颗粒物样品的高通量测序与数据统计分析总悬浮颗粒物中全部细菌的DNA测序所用扩增引物为V3+V4区338F/806R(5′-ACTCCTACGGGAGGCAGCA-3′/5′GGACTACHVGGG TWTCTAAT-3);PCR扩增程序如下:95 ℃预变性3 min;95 ℃变性30 s, 55 ℃退火30 s, 72 ℃延伸45 s, 27个循环;72 ℃延伸10 min;最后保持在10 ℃直到取出样品.扩增产物采用2.0%琼脂糖凝胶电泳检测, 使用AxyPrep DNA凝胶抽提试剂盒(AXYGEN)对PCR产物切胶纯化回收.
DNA序列通过Illumina Miseq平台进行测序, 按照97%相似性对非重复序列(不含单序列)进行操作分类单元OTU(operational taxonomic units)聚类, 在聚类过程中去除嵌合体, 得到OTU的代表序列.为了得到每个OTU对应的物种分类信息, 采用Ribosomal Database Project(RDP) Classifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析, 并分别在门(phylum)、纲(class)、目(order)、科(family)、属(genus)不同分类水平下统计各样本的群落组成.
2.3.3 可培养细菌克隆文库的构建与多样性分析可培养细菌DNA产物经过PCR扩增后构建克隆文库, 所用引物及扩增程序、覆盖度和多样性的计算参考本课题组前期报道(Han et al., 2013), 其中, 覆盖度C的计算方法如式(1)所示, C值≥75%方可认为挑取的克隆数足以反映真实的群落结构, C值越高, 表明挑取的克隆对真实群落的覆盖程度越高.
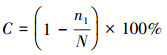
|
(1) |
式中, N为16S rDNA克隆文库的库容, 即挑取的阳性克隆总数目;n1为16S rDNA克隆文库中仅出现一次的克隆.
多样性以Shannon-Wiener指数进行评估, 计算方法如式(2)所示:
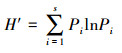
|
(2) |
式中, Pi为第i个OTU在克隆文库库容中所占的比例, 本研究中在相似度≥97%分类水平下划分OTU, OTU越多表明鉴定的细菌种类越多;S为克隆文库中OTU的总数目.
3 结果与分析(Results and analysis) 3.1 细菌气溶胶丰富度和多样性分析分别对污水厂不同采样点样品的克隆文库和高通量测序的结果进行统计, 结果见图 1.通过克隆文库挑取的阳性克隆数范围为43~107个, 划分的OTUs个数为11~37个;高通量测序技术获得的序列数范围为23656~42383个, 划分的OTUs个数为1212~1638个.基于克隆文库技术用某一阳性克隆的序列来代表某一物种、高通量测序技术用某一序列代表某一物种的原理, 显而易见, 各采样点用“总悬浮颗粒物采样器+高通量测序技术”检测出的物种数量均远多于用“Andersen六级采样器+克隆文库技术”检测出的物种数量, 表明高通量测序技术对于细菌气溶胶群落结构丰富度的表征具有优势.
 |
| 图 1 16S rDNA克隆文库(a)和高通量测序技术(b)分析细菌气溶胶获取的测序信息 Fig. 1 Sequencing information of airborne bacteria analyzed by 16S rDNA clone library (a) and high-throughput sequencing(b) |
进一步对上述测序结果做细菌群落组成覆盖度的评价, 结果如图 2所示.克隆文库技术对污水处理厂不同采样点气溶胶中细菌的覆盖度范围为77.50%~90.40%, 不同采样点气溶胶中细菌群落结构的覆盖度差别较大;而高通量测序对所有细菌气溶胶的覆盖度均大于99%.上述结果表明, 尽管克隆文库技术和高通量测序技术在呈现气溶胶中细菌群落结构覆盖度方面均达到了实验理论的标准要求, 但高通量测序技术在呈现群落结构覆盖度的稳定性和全面性方面显著优于克隆文库技术.
 |
| 图 2 通过16S rDNA克隆文库和高通量测序技术分析细菌气溶胶群落结构的覆盖度 Fig. 2 Coverage of airborne bacteria community structure sequenced by 16S rDNA clone library and high-throughput sequencing |
对各采样点气溶胶样品中的细菌多样性(属分类水平)进行分析, 结果见图 3.依据式(2)计算的气溶胶中的细菌Shannon-Wiener指数表明, 克隆文库分析可培养细菌的多样性指数为0.58(粗格栅)~2.37(污泥脱水间), 而通过高通量测序对全部细菌多样性指数的分析结果为5.06(细格栅)~5.99(污泥脱水间).高通量测序技术检测到的各采样点气溶胶中的细菌多样性均远高于克隆文库检测到的结果.
 |
| 图 3 通过16S rDNA克隆文库和高通量测序技术分析细菌气溶胶的Shannon-Wiener指数 Fig. 3 Shannon-Wiener index for airborne bacteria analyzed by 16S rDNA clone library and high-throughput sequencing |
在门、纲、目、科和属5种分类水平下, 分别对污水处理厂各采样点的细菌气溶胶克隆文库和高通量测序的结果进行统计(表 1), 结果表明, 无论在何种分类水平下, 高通量测序对于气溶胶中的细菌群落组成的解析更全面.
| 表 1 克隆文库和高通量测序对细菌气溶胶在不同分类水平的对比 Table 1 Taxon comparison of clone library and high-throughput sequencing |
图 4展示的是在门分类水平下, “总悬浮颗粒物采样器+高通量测序技术”和“Andersen六级采样器+克隆文库技术”对细菌气溶胶群落组成的解析结果.在所有采样点的细菌气溶胶中, 克隆文库仅检测到厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)和放线菌门(Actinobacteria)3种细菌门类, 其中前两者为主要的细菌门类(图 4a), 同时, 这两种菌门在不同采样点的分布差别较大, 例如, 厚壁菌门在粗格栅(90.0%)、细格栅(92.2%)和沉砂池(78.9%)的占比较高, 而变形菌门在曝气池水面(62.1%)和污泥脱水间(74.4%)为优势菌门.
 |
| 图 4 通过16S rDNA克隆文库(a)和高通量测序技术(b)分析细菌气溶胶在门类水平的群落结构 Fig. 4 Community structure of airborne bacteria on phylum level analyzed by 16S rDNA clone library (a) and high-throughput sequencing technology(b) |
高通量测序获得的细菌门类共有34个(表 1), 其中, 丰度≥1%的有11个, 其100%地覆盖了克隆文库测得的3种优势菌门.除此之外, 还特异地鉴定到包括拟杆菌门(Bacteroidetes, 3.06%~13.48%)等在内的其他优势菌门, 总比例为72.1%~91.7%(图 4b).
上述结果表明, 在门分类水平下, 克隆文库技术由于测序通量低和挑取阳性克隆的随机性等弊端(张玉等, 2018), 重复检出厚壁菌门和变形菌门细菌, 高估了其在污水厂细菌气溶胶中的占比, 而低估甚至忽略了包括拟杆菌门、绿弯菌门(Chloroflexi)、酸杆菌门(Acidobacteria)等在内的其他细菌门类的分布比例(图 4b).高通量测序可一次并行对几十万到几百万条DNA分子进行序列测定, 克服了克隆文库测序的上述弊端, 可以更全面、客观地对微生物群落多样性进行评估.
在属分类水平下, 利用克隆文库分析技术检测气溶胶中的细菌群落组成如图 5所示, 在鉴定到的31个细菌菌属中(丰度均大于1%), 各个采样点的群落组成差别较大, 例如, 芽孢杆菌属(Bacillus)为所有采样点的优势菌属, 其中, 在粗格栅和细格栅的细菌气溶胶群落组成中占绝对优势, 分别占87.0%和74.8%, 而在曝气池水面和污泥脱水间的比例分别为6.90%和15.0%;气单胞菌属(Aeromonas)为曝气池水面处的优势菌属(55.2%).导致不同采样点气溶胶中细菌群落结构差异的原因一方面是由于上述克隆文库的技术弊端;另一方面与污水厂不同处理工段的构筑物特点、污水处理设施等有关, 例如, 本研究选取的这座污水厂的粗格栅和细格栅为室内操作间, 其中, 粗格栅为地下7 m左右的操作间, 粗格栅和细格栅均不加盖, 湍急的进水将污水中大量的微生物释放到空气中形成气溶胶, 在具有稳定气象条件的室内环境中不断累积, 导致其群落组成与设置于室外曝气池处的气溶胶中的细菌群落组成明显不同.
 |
| 图 5 通过16S rDNA克隆文库技术分析细菌气溶胶在属分类水平的群落结构 Fig. 5 Community structure of airborne bacteria analyzed by 16S rDNA clone library at genus level |
高通量测序技术共检测到422个细菌菌属(表 1), 其中丰度≥1%的共有79个(丰度小于1%的全部归类为Others)(图 6).两种测序技术共同鉴定到6个菌属, 高通量测序技术特异地检测到的细菌菌属占83.3%~98.5%, 其对于群落多样性的解析显著优于克隆文库技术.
 |
| 图 6 高通量测序技术分析细菌气溶胶在属分类水平的群落结构 Fig. 6 Community structure of airborne bacteria analyzed by high throughout sequencing at genus level |
值得一提的是, “总悬浮颗粒物采样器+高通量测序技术”和“Andersen六级采样器+克隆文库技术”对同一采样点细菌气溶胶的全部细菌和可培养细菌的优势群落鉴定结果几乎完全不同, 而高通量测序对全部细菌群落结构的解析更能证明污水厂不同采样点细菌气溶胶与其逸散源的关系, 例如, 在进水段的粗格栅和细格栅处的优势菌属为气单胞菌属(10.7%、12.5%)和消化链球菌属(Peptostreptococcaceae_unclassified)(15.1%、11.1%), 前者普遍存在于淡水、淤泥和污水等环境中(Parker et al., 2011), 而后者常见于人和动物的口腔、肠道和呼吸道, 在人类女性泌尿生殖道也是正常的生理菌群, 污水厂的进水作为收纳这类微生物的水体(Lu et al., 2015;Jiang et al., 2015), 进水过程中的跌水等环节会将其释放到周围的环境中(Brandi et al., 2000;Li et al., 2013);而污泥脱水间气溶胶中7.1%的细菌隶属于细菌菌胶团(Zoogloea), Zoogloea为活性污泥的主体, 而本研究中污泥脱水间内的污泥脱水机为离心式且为不加盖的设备, 在脱水过程中会将活性污泥中的细菌直接释放到周围空气中.
上述结果表明, 细菌气溶胶直接提取DNA并通过高通量测序对细菌气溶胶优势菌的解析, 更能说明细菌气溶胶与其释放源的关联性, 可更全面、真实地反映细菌气溶胶的群落分布状态, 近几年高通量测序技术的快速发展, 为空气微生物的宏基因组解析提供了操作简便、通量大、信息全面、经济适用的技术支持(Behzad et al., 2015), 但该分析方法由于读长较短, 对于准确鉴定细菌的种类存在一定缺陷.而通过传统的可培养法结合克隆文库收集并解析细菌气溶胶, 实际是选择性地收集易于培养细菌的过程, 因而导致其分析结果具有较强的随机性和较大偏差, 此外, 该法操作繁复, 大批量、复杂空气微生物的解析需大量的人力和高昂的费用, 获取的序列通量有限, 尽管如此, 16S rDNA克隆文库作为一项成熟的测序技术, 可操作性强, 获得的测序片段较长(大约为1500 bp), 可以准确地确定微生物的分类, 对微生物多样性较低样本的准确鉴定依然适用.
4 结论(Conclusions)通过总悬浮颗粒物采样器收集污水处理全过程中的细菌气溶胶, 并通过高通量测序技术对其进行序列测定, 结果表明, 相对于对照组采用Andersen采样器的传统可培养方法和克隆文库分析方法, 前者特异地检测到细菌气溶胶中83.3%~98.5%的细菌菌属, 气溶胶中的多个细菌优势菌属在污水、污泥中广泛存在, 因此, 采用“总悬浮颗粒物采样器+高通量测序技术”在解析污水处理厂气溶胶中细菌多样性、群落结构覆盖度、稳定性和全面性方面具有明显的优势, 能客观说明细菌气溶胶与其释放源的关联性, 全面和真实地反映细菌气溶胶的群落分布状态.但该分析方法由于读长较短, 对于准确鉴定细菌的种类尚存在一定缺陷.
Behzad H, Gojobori T, Mineta K. 2015. Challenges and opportunities of airborne metagenomics[J]. Genome Biology and Evolution, 7(5): 1216–1226.
DOI:10.1093/gbe/evv064
|
Brandi G, Sisti M, Amagliani G. 2000. Evaluation of the environmental impact of microbial aerosols generated by wastewater treatment plants utilizing different aeration systems[J]. Journal of Applied Microbiology, 88(5): 845–852.
DOI:10.1046/j.1365-2672.2000.01024.x
|
Cao C, Jiang W, Wang B, et al. 2014. Inhalable microorganisms in Beijing's PM2.5 and PM10 pollutants during a severe smog event[J]. Environmental Science and Technology, 48: 1499–1507.
DOI:10.1021/es4048472
|
Dannemiller K C, Lang-yona N, Yamamoto N, et al. 2014. Combining real-time PCR and next-generation DNA sequencing to provide quantitative comparisons of fungal aerosol populations[J]. Atmospheric Environment, 84: 113–121.
DOI:10.1016/j.atmosenv.2013.11.036
|
Despr S V R, Alex H J, Burrows S M, et al. 2012. Primary biological aerosol particles in the atmosphere:a review[J]. Tellus Series B-Chemical and Physical Meteorology, 64: 15598.
DOI:10.3402/tellusb.v64i0.15598
|
Dong L, Qi J, Shao C, et al. 2016. Concentration and size distribution of total airborne microbes in hazy and foggy weather[J]. Science of the Total Environment, 541: 1011–1018.
DOI:10.1016/j.scitotenv.2015.10.001
|
方治国, 郝翠梅, 姚文冲, 等. 2016. 空气微生物群落解析方法:从培养到非培养[J]. 生态学报, 2016, 36(14): 4244–4253.
|
方治国, 欧阳志云, 胡利锋, 等. 2005. 北京市三个功能区空气微生物中值直径及粒径分布特征[J]. 生态学报, 2005, 35(12): 3220–3224.
DOI:10.3321/j.issn:1000-0933.2005.12.015 |
房文艳, 高新磊, 李继, 等. 2015. 城市社区农贸市场空气微生物及抗生素抗性基因研究[J]. 生态毒理学报, 2015, 10(5): 95–99.
|
Han Y, Li L, Liu J, et al. 2012. Microbial structure and chemical components of aerosols caused by rotating brushes in a wastewater treatment plant[J]. Environmental Science and Pollution Research, 19(9): 4097–4108.
DOI:10.1007/s11356-012-0885-1
|
Han Y P, Li L, Liu J X. 2013. Characterization of the airborne bacteria community at different distances from the rotating brushes in a wastewater treatment plant by 16S rRNA gene clone libraries[J]. Journal of Environmental Sciences-China, 25(1): 5–15.
DOI:10.1016/S1001-0742(12)60018-7
|
Jiang W J, Liang P, Wang B Y, et al. 2015. Optimized DNA extraction and metagenomic sequencing of airborne microbial communities[J]. Nature Protocols, 10(5): 768–779.
DOI:10.1038/nprot.2015.046
|
Kumaraswamy R, Amha Y M, Anwar, et al. 2014. Molecular analysis for screening human bacterial pathogens in municipal wastewater treatment and reuse[J]. Environmental Science and Technology, 48: 11610–11619.
DOI:10.1021/es502546t
|
Lange J L, Thorne P S, Lynch N. 1997. Application of flow cytometry and fluorescent in situ hybridization for assessment of exposures to airborne bacteria[J]. Appllied and Environmental Microbiology, 63(4): 1557–1563.
|
Li Y P, Yang L W, Meng Q L, et al. 2013. Emission characteristics of microbial aerosols in a municipal sewage treatment plant in Xi'an, China[J]. Aerosol and Air Quality Research, 13(1): 343–349.
DOI:10.4209/aaqr.2012.05.0123
|
Lu X, Zhang X X, Wang Z, et al. 2015. Bacterial pathogens and community composition in advanced sewage treatment systems revealed by metagenomics analysis based on high-throughput sequencing[J]. PLoS One, 10(5): e0125549.
DOI:10.1371/journal.pone.0125549
|
李涛. 2003. 空气微生物采样及发展趋势[J]. 中国卫生检验杂志, 2003, 13(5): 538–539.
DOI:10.3969/j.issn.1004-8685.2003.05.003 |
刘建伟, 张俊超, 马文林, 等. 2013. 城市污水处理厂微生物气溶胶污染和粒径分布特征[J]. 生态环境学报, 2013, 22(4): 657–661.
DOI:10.3969/j.issn.1674-5906.2013.04.018 |
Parker J L, Shaw J G. 2011. Aeromonas spp.clinical microbiology and disease[J]. Journal of Infection, 62(2): 109–118.
DOI:10.1016/j.jinf.2010.12.003
|
Peccia J, Hernandez M. 2006. Incorporating polymerase chain reaction-based identification, population characterization, and quantification of microorganisms into aerosol science:A review[J]. Atmospheric Environment, 40(21): 3941–3961.
DOI:10.1016/j.atmosenv.2006.02.029
|
Urbano R, Palenik B, Gaston C J, et al. 2011. Detection and phylogenetic analysis of coastal bioaerosols using culture dependent and independent techniques[J]. Biogeosciences, 8(2): 301–309.
DOI:10.5194/bg-8-301-2011
|
Wery N, Lhoutellier C, Ducray F, et al. 2008. Behaviour of pathogenic and indicator bacteria during urban wastewater treatment and sludge composting, as revealed by quantitative PCR[J]. Water Research, 42(1/2): 53–62.
|
Xu Z, Yao M. 2013. Monitoring of bioaerosol inhalation risks in different environments using a six-stage Andersen sampler and the PCR-DGGE method[J]. Environmental Monitoring and Assessment, 185(5): 3993–4003.
DOI:10.1007/s10661-012-2844-1
|
Yamamoto N, Nazaroff W W, Peccia J. 2014. Assessing the aerodynamic diameters of taxon-specific fungal bioaerosols by quantitative PCR and next-generation DNA sequencing[J]. Journal of Aerosol Science, 78: 1–10.
DOI:10.1016/j.jaerosci.2014.08.007
|
于玺华. 2002. 现代空气微生物学[M]. 北京: 人民军医出版社.
|
张玉, 米铁柱, 甄毓, 等. 2018. 海洋沉积物中硫酸盐还原菌和硫氧化菌群落分析方法的比较[J]. 环境科学, 2018, 39(1): 1–17.
|