 2018, Vol. 38
2018, Vol. 38



近年来, 基于过一硫酸盐(PMS)的化学氧化工艺在地下水原位修复和非原位修复中的应用越来越受到关注(Cui et al., 2016; Chen et al., 2018).PMS是通过利用自由基驱动的过程或直接电子转移来处理各种各样的污染物, 研究表明(Qi et al., 2017; Deng et al., 2017; Jaafarzadeh et al., 2016), PMS能够降解高毒性和持久性有机物, 且与其他氧化剂相比价格便宜.常温下, PMS在固态下非常稳定, 在纯水中能够稳定保持数月, 很难分解, 通过紫外光照射(Gao et al., 2012; He et al., 2013)、加热(Waldemer et al., 2007; Liang et al., 2008)、超声波(Chen et al., 2012)、过渡金属离子(Liang et al., 2004; Kusic et al., 2011; Rodriguez et al., 2014)等手段, 能够活化PMS, 产生具有强氧化性的活性物质, 如硫酸根自由基(SO4-·)、羟基自由基(·OH)等.其中, 使用紫外光、加热和超声波的方式产生的能耗大、操作复杂, 而过渡金属活化会导致金属的浸出从而造成二次污染, 这些活化方式的缺陷都限制了它们在实际工程的应用.
传统的高级氧化工艺(AOP)在去除新兴污染物方面表现出良好的性能, 其机理主要取决于·OH(Wang et al., 2012; Wang et al., 2016)和SO4-· (Zhao et al., 2017).·OH是一种非选择性的强氧化剂, 氧化还原电位为2.8 V, 可以破坏有机污染物的结构, 甚至在一定程度上使其矿化(Wang et al., 2016).目前, 几乎所有活化PMS的体系都以产生SO4-·为主要活性物种, 基于SO4-·的高级氧化工艺被公认为降解难处理有机污染物的有效方法(Oh et al., 2016).与·OH相比, SO4-·具有相同或甚至更高的氧化还原电位(2.5~3.1 V).然而, 这种自由基制方法可能会促成自由基与有机底物发生反应, 这样的反应会不可避免地形成副产物, 如在含有痕量溴离子杂质的有机废水中, 采用自由基制方法会产生溴代芳烃副产物, 具有致癌的危害(Hu et al., 2016; Fang et al., 2017).因此, 基于活化PMS的非自由基机制降解污染物的方法引起了广泛关注, 已有研究表明(Zhou et al., 2017), 酮基活化PMS的非自由基制方法产生的氧化物种为单线态氧分子, 对污染物的选择性较高, 受周围环境的影响, 能够不断产生和淬灭, 且不会产生毒害副产物.因此, 研究丙酮/PMS体系中非自由基氧化过程, 具有重要的意义.本文通过丙酮活化PMS对偶氮染料AO7进行降解, 分析其可能存在的活化机理, 研究丙酮投加量、PMS浓度、初始pH、共存阴离子、腐殖酸浓度等因素对AO7降解的影响, 并对各反应条件下的降解反应进行分析.
2 材料与方法(Materials and methods) 2.1 材料试剂丙酮、酸性橙7(AO7)购于国药集团化学有限公司;过一硫酸盐(KHSO5·0.5KHSO4·0.5K2SO4, PMS)购于Sigma-Aldrich;9, 10-二苯基蒽(9, 10-DPA)购于阿拉丁生化试剂有限公司.实验所用亚硝酸钠(NaNO2)、碘化钾(KI)、氯化钠(NaCl)、碳酸钠(Na2CO3)、碳酸氢钠(NaHCO3)、甲醇(CH3OH)、叔丁醇(C4H10O, TBA)、糠醇(C5H6O2, FFA)、氢氧化钠(NaOH)、硫酸(H2SO4)均为分析纯, 购于国药集团化学试剂有限公司.实验用水为超纯水.
2.2 降解实验室温下, 将一定浓度的PMS储备液注入100 mL的锥形瓶中, 同时往锥形瓶中加入一定量的超纯水, 并用稀H2SO4或NaOH调节pH值;然后迅速加入一定量的丙酮和一定量的AO7溶液, 使得总溶液达到100 mL, 采用磁力搅拌混合启动反应.间隔一定时间取样, 迅速加入过量NaNO2, 猝灭PMS, 终止反应, 待后续测定.
2.3 分析方法使用Mapada UV-1600(PC)紫外可见分光光度计, 于AO7最大吸收波长484 nm处测定样品的吸光度, 代入标准曲线求得浓度C.使用Mapada UV-1600(PC)紫外可见分光光度计于352 nm处测定PMS剩余浓度.
3 结果与讨论(Results and discussion) 3.1 不同初始条件对AO7降解性能的影响不同反应体系中AO7的降解效果如图 1所示.由图 1可知, 在30 min内, 丙酮单独反应体系对AO7的降解效果不明显, 几乎不脱色, PMS单独氧化体系中AO7的脱色率为60.4%, 然而在丙酮/PMS体系中, AO7的脱色率明显增加, 达到99.8%.由此得出, PMS单独氧化体系对AO7的降解效果欠佳, 丙酮单独体系对AO7无降解作用, 但丙酮/PMS体系能够快速降解AO7, 大大地缩短了染料降解的时间.
 |
| 图 1 不同初始条件对AO7降解的影响(pH=9.0, T=298 K, [AO7]=20 mg·L-1, [PMS]=1 mmol·L-1, [丙酮]=10 mmol·L-1) Fig. 1 Effect of different initial conditions on the degradation of AO7 |
在碱性条件下, PMS单独氧化体系中, PMS被碱活化产生超氧自由基(O2-·)和单线态氧(1O2)(Qi et al., 2016), 对AO7有一定的降解作用.单独丙酮时, 溶液中不产生氧化性物质, 因此, AO7不会被降解.在丙酮/PMS体系中, 丙酮和过一硫酸盐作用, 可能产生活性物种, 加快了AO7的降解速率.
3.2 丙酮对PMS的活化机理为了验证丙酮/PMS体系降解AO7的机理, 向反应体系中加入自由基猝灭剂判断起作用的自由基(Oh et al., 2015).本实验采用SO4-·、·OH猝灭剂甲醇(MeOH)、·OH猝灭剂叔丁醇(TBA)和单线态氧猝灭剂糠醇(FFA)(Haag et al., 1984)对反应体系进行自由基鉴定, 加入的甲醇、叔丁醇和糠醇浓度为1 mol·L-1, 降解结果如图 2所示.由图 2可知, 在50 min时, 不加猝灭剂的体系中AO7的降解率为99.6%;向丙酮/PMS体系中加入甲醇和TBA, 发现AO7的降解没有受到明显抑制, 其脱色率分别是99.3%、99.9%;然而, 往丙酮/PMS体系中加入FFA时, 50 min时AO7的脱色率仅为2.0%.
 |
| 图 2 MeOH、TBA和FFA对丙酮/PMS体系降解AO7的影响(pH=9.0, T=298 K, [AO7]=20 mg·L-1, [PMS]=1 mmol·L-1, [丙酮]=10 mmol·L-1) Fig. 2 Effect of methanol, TBA and FFA on the degradation of AO7 in acetone/PMS system |
该体系中AO7的降解率不会受到MeOH和TBA的抑制, 由此说明丙酮活化PMS没有产生SO4-·和·OH.然而, 在FFA淬灭剂的作用下, AO7的脱色效果迅速下降, 脱色反应几乎完全被抑制, 由此说明反应体系中可能产生了1O2, 1O2可能是该反应体系的主要氧化物种.有研究表明(吕庆銮等, 2008), 可以采用9, 10-DPA对1O2进行化学捕获, 9, 10-DPA能够快速与单线态氧反应生成稳定的内氧化物(式(1)), 反应常数为1.3×106 L·mol-1·s-1, 通过紫外分光光度计测定DPA在355 nm的吸光度的减少量即可以测量单线态氧的含量.因此, 为了验证上述猜想, 利用9, 10-DPA对反应过程中1O2进行化学捕获.从图 3可以看出, 在355 nm处的吸光度由0.4499下降至0.3420, 证实反应体系中产生了1O2.

|
(1) |
 |
| 图 3 9, 10-DPA反应前后紫外可见光谱(pH=9.0, T=298 K, [PMS]=1 mmol·L-1, [丙酮]=10 mmol·L-1) Fig. 3 UV-visible spectra of 9, 10-DPA before and after reaction |
丙酮投加量对AO7降解效果的影响如图 4a所示, 其中, 固定PMS的投加量为1 mmol·L-1, 25 min时PMS单独氧化体系中AO7的脱色率为60.0%, 向反应体系中加入丙酮, 投加量为1 mmol·L-1时, AO7的降解率达到76.9%;当投加量增加到20 mmol·L-1时, 丙酮/PMS体系内AO7的降解率达到100.0%.由此可知, 随着丙酮投加量的增大, AO7的降解速度加快, 降解周期缩短.这一结果表明, 随着丙酮投加量增加, 催化剂提供的活化点位增加, 促进PMS产生大量的1O2, 促使活化效果增强.
 |
| 图 4 丙酮投加量对降解AO7的影响(pH=9.0, T=298 K, [AO7]=20 mg·L-1, [PMS]=1 mmol·L-1) Fig. 4 Effect of acetone dosage on the degradation of AO7 |
催化剂用量分别为0、1、5、10、15、20 mmol·L-1时, AO7的降解速率kobs值分别为0.040、0.055、0.093、0.180、0.293和0.316 min-1.显然, AO7降解速率随着催化剂投加量的增加而增加.综合考虑经济成本和降解速率, 选择10 mmol·L-1作为最佳用量.
3.4 PMS浓度的影响不同浓度PMS对AO7降解效果的影响如图 5所示, 其中, 固定丙酮投加量为10 mmol·L-1.由图 5可知, 与丙酮投加量对AO7降解效果影响的变化趋势相似, PMS浓度越大, AO7的降解速率越快, 完全降解所需的时间越短.当PMS浓度为0.5 mmol·L-1时, 反应至30 min时AO7的脱色率为81.6%;当PMS的浓度增大至1 mmol·L-1, AO7的脱色率达到99.8%;当PMS的浓度增大至2 mmol·L-1, AO7在20 min时完全降解;继续增大PMS的浓度至10 mmol·L-1, AO7在8 min时完全降解.主要原因可能是当活化剂丙酮用量为10 mmol·L-1时, 催化剂表面活化点位仍未被PMS完全占据, 提高PMS浓度会使丙酮活化PMS产生中间体1O2的量增加, 更有利于促进对偶氮染料AO7的氧化降解, 大大缩短降解时间.而随着PMS浓度的进一步增加, 相应的催化剂比例下降, 而过量的PMS会在碱性条件下活化产生1O2和O2-·, 大大缩短偶氮染料降解的时间, 造成PMS的浪费, 应在提高降解速率的同时, 适当考虑经济因素, 将PMS浓度控制在适当范围内, 后续实验选取PMS浓度为1 mmol·L-1.
 |
| 图 5 不同PMS浓度对降解AO7的影响(pH=9.0, T=298 K, [AO7]=20 mg·L-1, [丙酮]=10 mmol·L-1) Fig. 5 Effect of different PMS concentrations on the degradation of AO7 |
不同初始pH值对丙酮/PMS反应活化体系降解AO7的影响如图 6所示.由图可知, 反应初始pH分别为5.0、6.0、7.0、8.0、9.0、10.0时, 25 min时丙酮/PMS体系对AO7的脱色率分别为8.6%、8.4%、12.1%、40.4%、99.8%、100.0%;随着pH值的减小, 反应速率逐渐降低, 在酸性pH(5.0、6.0)条件下, AO7的降解速率较低, 几乎不降解.
 |
| 图 6 不同初始pH对AO7降解的影响(T=298K, [AO7]=20 mg·L-1, [丙酮]=10 mmol·L-1, [PMS]=1 mmol·L-1) Fig. 6 Effect of different initial pH on the degradation of AO7 |
周扬等(2015)在苯醌活化PMS降解磺胺甲恶挫(SMX)的研究中发现, pH由7升至10, SMX的降解速率随之增加.自由基淬灭研究表明, 反应体系中不产生自由基, 主要氧化物种为1O2, 并提出相应的反应机制, 即PMS与苯醌之间形成二环氧乙烷中间体, 随后该中间体分解形成1O2, 主要作用于苯醌的羰基.丙酮与苯醌拥有结构类似的羰基, 碱性条件下, AO7的降解速率较快, 由此可以提出合理的反应机制, 过一硫酸盐在碱性条件下的存在形式不稳定, 更容易被活化, OH-会与HSO5-反应, 并且在碱性条件下, 丙酮活化PMS快速产生环氧中间体, 中间体迅速分解产生1O2(式(2)~(6));同时, 在pH=9.0的条件下, 对单独PMS和丙酮/PMS体系分别进行PMS剩余浓度的测定, 单独PMS体系中PMS的剩余浓度变化微小, 丙酮/PMS体系中PMS的剩余浓度不断下降(图 7), 说明该体系中是丙酮参与了PMS的活化.酸性条件下, 反应体系中的H+会与HSO5-反应形成氢键(房聪等, 2018), 反应会被钝化, 因此, 不能有效地活化PMS.
 |
| 图 7 体系PMS剩余浓度(pH=9.0, T=298K, [丙酮]=10 mmol·L-1, [PMS]=1 mmol·L-1) Fig. 7 Residual concentration of PMS in the system |
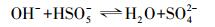
|
(2) |
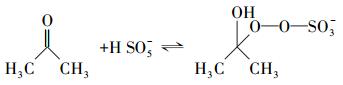
|
(3) |
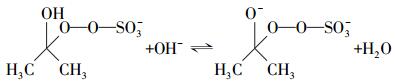
|
(4) |
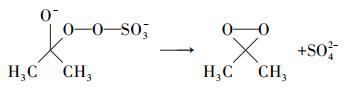
|
(5) |
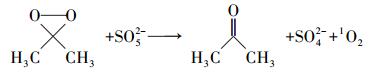
|
(6) |
Cl-和CO32-对丙酮/PMS反应体系氧化降解AO7的影响如图 8所示.在20 min时, 丙酮/PMS体系中AO7的降解率为97.6%;Cl-/PMS体系中AO7的降解率为84.3%;CO32-/PMS体系中AO7的降解率为81.5%;单独PMS体系中AO7的降解率仅为48.4%.然而, 当n(Cl-):n(丙酮)=1:1时, AO7在18 min时就可以完全降解;当n(CO32-):n(丙酮)=1:1时, 在20 min时AO7完全降解.
 |
| 图 8 Cl-和CO32-对丙酮/PMS体系降解AO7的影响(pH=9.0, T=298 K, [PMS]=1 mmol·L-1, [丙酮]=10 mmol·L-1, [Cl-]=10 mmol·L-1, [CO32-]=10 mmol·L-1, [AO7]=20 mg·L-1) Fig. 8 Effect of Cl- and CO32- on the degradation of AO7 in the acetone/PMS system |
可以看出, 在丙酮/PMS体系中, Cl-的加入显著促进了AO7的降解, 缩短了降解时间.主要原因是Cl-与PMS产生的HSO5-自由基反应生成氧化性较强的ClO-(式(7)).同时, 在丙酮的催化氧化和Cl-的氯化作用下, 体系中发生的反应会更加复杂.在1O2和ClO-自由基共同作用下, 能够快速地降解AO7.
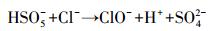
|
(7) |
当向丙酮/PMS体系中加入CO32-时, 对AO7的降解也有促进作用, 但促进效果不显著.主要原因可能是CO32-的加入增强了离子强度, 会加剧染料之间的聚合, 从而促进AO7的降解.
3.7 腐殖酸浓度对AO7降解速率的影响天然有机物质(NOM)一直存在于天然水体中, Deng等(2017)发现它们对高级氧化工艺有显著影响.因此, 本文研究了不同腐殖酸(HA)浓度对丙酮/PMS体系氧化降解AO7的影响, 结果如图 9所示.HA浓度分别为0、5、10、15、20 mg·L-1时, 40 min时AO7的降解率分别为100.0%、99.2%、98.3%、97.2%、96.5%.结果表明, HA的加入对反应体系有不明显的抑制作用, 这可以通过这些反应部位在NOM分子中竞争性清除自由基来解释, 由于丙酮/PMS活化体系通过产生1O2来攻击降解偶氮染料, 几乎不产生自由基, 因此, HA的加入对反应体系的抑制不明显.
 |
| 图 9 HA浓度对丙酮/PMS体系降解AO7的影响(pH=9.0, T=298 K, [AO7]=20 mg·L-1, [PMS]=1 mmol·L-1, [丙酮]=10 mmol·L-1) Fig. 9 Effect of HA concentration on the degradation of AO7 in the acetone/PMS system |
图 10为AO7在丙酮/PMS体系中降解的紫外可见光谱.由图 10可知, AO7的光谱扫描主要有两处特征吸收峰, 分别在484 nm和310 nm处.根据文献(王莹等, 2017)可知, 484 nm处对应的是AO7的发色基团偶氮键, 310 nm处对应的是AO7的萘环结构.当pH=9.0时, 随着反应体系的活化, AO7在484 nm和310 nm两处的波峰强度都在不断减小, 在30 min时, 两处的波峰接近消失, 根据研究,354 nm处为抑制剂的锋, 这说明偶氮染料中的偶氮键和萘环结构不断被氧化物质1O2破坏, 从而得到降解.
 |
| 图 10 AO7降解的紫外可见光谱(pH=9.0, T=298 K, [AO7]=20 mg·L-1, [PMS]=1 mmol·L-1, [丙酮]=10 mmol·L-1) Fig. 10 UV-visible spectra for degradation of AO7 |
1) 丙酮活化PMS氧化降解AO7的效果良好, 可以通过淬灭实验证实丙酮活化PMS产生的具有氧化性的活性物种主要是单线态氧1O2.
2) 随着丙酮投加量、PMS浓度及共存阴离子浓度的升高, AO7的降解速率也随之加快.在碱性条件下更有利于丙酮活化PMS, 从而加快AO7的降解速率.腐殖酸的加入对AO7的降解有不明显的抑制作用.
3) 通过对丙酮/PMS体系降解AO7的紫外可见扫描光谱分析可知, AO7降解过程中在484 nm和310 nm处吸收峰强度明显减小, 表明AO7分子中发色基因偶氮键及萘环结构均在一定程度上被破坏, 从而达到降解的目的.
Chen J, Fang C, Xia W, et al. 2018. Selective transformation of β-Lactam antibiotics by peroxymonosulfate:reaction kinetics and non-radical mechanism[J]. Environmental Science & Technology, 52(3): 1461–1470.
|
Chen W S, Su Y C. 2012. Removal of dinitrotoluenes in wastewater by sono-activated persulfate[J]. Ultrasonics Sonochemistry, 19(4): 921–927.
DOI:10.1016/j.ultsonch.2011.12.012
|
Cui J, Zhang L, Xi B, et al. 2016. Chemical oxidation of benzene and trichloroethylene by a combination of peroxymonosulfate and permanganate linked by in-situ generated colloidal/amorphous MnO2[J]. Chemical Engineering Journal, 313: 815–825.
|
Deng J, Ge Y, Tan C, et al. 2017. Degradation of ciprofloxacin using α-MnO2 activated peroxymonosulfate process:Effect of water constituents, degradation intermediates and toxicity evaluation[J]. Chemical Engineering Journal, 330: 1390–1400.
DOI:10.1016/j.cej.2017.07.137
|
Deng L, Shi Z, Zou Z, et al. 2017. Magnetic EDTA functionalized CoFe2O4 nanoparticles (EDTA-CoFe2O4) as a novel catalyst for peroxymonosulfate activation and degradation of Orange G[J]. Environmental Science & Pollution Research, 24(12): 1–13.
|
Fang C, Wang Z, Feng M, et al. 2017. Trace bromide ion impurity leads to formation of chlorobromoaromatic by-products in peroxymonosulfate-based oxidation of chlorophenols[J]. Chemosphere, 182: 624–629.
DOI:10.1016/j.chemosphere.2017.05.065
|
房聪, 房烽, 张黎明, 等. 2018. 秸秆活性炭活化过一硫酸盐降解酸性橙7[J]. 环境科学学报, 2018, 38(1): 242–250.
|
Gao Y, Gao N, Deng Y, et al. 2012. Ultraviolet (UV) light-activated persulfate oxidation of sulfamethazine in water[J]. Chemical Engineering Journal, 195-196: 248–253.
DOI:10.1016/j.cej.2012.04.084
|
Haag W R, Hoigne J R, Gassman E, et al. 1984. Singlet oxygen in surface waters-Part I:Furfuryl alcohol as a trapping agent[J]. Chemosphere, 13(5): 631–640.
|
He X, Cruz A A D L, Dionysiou D D. 2013. Destruction of cyanobacterial toxin cylindrospermopsin by hydroxyl radicals and sulfate radicals using UV-254 nm activation of hydrogen peroxide, persulfate and peroxymonosulfate[J]. Journal of Photochemistry & Photobiology A Chemistry, 251(48): 160–166.
|
Hu P, Long M. 2016. Cobalt-catalyzed sulfate radical-based advanced oxidation:A review on heterogeneous catalysts and applications[J]. Applied Catalysis B Environmental, 181: 103–117.
DOI:10.1016/j.apcatb.2015.07.024
|
Jaafarzadeh N, Ghanbari F, Ahmadi M. 2016. Catalytic degradation of 2, 4-dichlorophenoxyacetic acid (2, 4-D) by nano-Fe2O3 activated peroxymonosulfate:Influential factors and mechanism determination[J]. Chemosphere, 169: 568–576.
|
Kusic H, Peternel I, Ukic S, et al. 2011. Modeling of iron activated persulfate oxidation treating reactive azo dye in water matrix[J]. Chemical Engineering Journal, 172(1): 109–121.
DOI:10.1016/j.cej.2011.05.076
|
吕庆銮, 张苗, 岳宁宁, 等. 2008. 单线态氧的检测及分析应用研究进展[J]. 化学分析计量, 2008, 17(3): 74–77.
DOI:10.3969/j.issn.1008-6145.2008.03.027 |
Liang C J, Bruell C J. 2008. Thermally activated persulfate oxidation of trichloroethylene:Experimental investigation of reaction orders[J]. Industrial & Engineering Chemistry Research, 47(9): 2912–2918.
|
Liang C, Bruell C J, Marley M C, et al. 2004. Persulfate oxidation for in situ remediation of TCE.I.Activated by ferrous ion with and without a persulfate-thiosulfate redox couple[J]. Chemosphere, 55(9): 1213–1223.
DOI:10.1016/j.chemosphere.2004.01.029
|
Oh W D, Dong Z, Lim T T. 2016. Generation of sulfate radical through heterogeneous catalysis for organic contaminants removal:Current development, challenges and prospects[J]. Applied Catalysis B Environmental, 194: 169–201.
DOI:10.1016/j.apcatb.2016.04.003
|
Oh W D, Lua S K, Dong Z, et al. 2015. Performance of magnetic activated carbon composite as peroxymonosulfate activator and regenerable adsorbent via sulfate radical-mediated oxidation processes[J]. Journal of Hazardous Materials, 284: 1–9.
DOI:10.1016/j.jhazmat.2014.10.042
|
Qi C, Liu X, Lin C, et al. 2017. Activation of peroxymonosulfate by microwave irradiation for degradation of organic contaminants[J]. Chemical Engineering Journal, 315: 201–209.
DOI:10.1016/j.cej.2017.01.012
|
Qi C, Liu X, Ma J, et al. 2016. Activation of peroxymonosulfate by base:Implications for the degradation of organic pollutants[J]. Chemosphere, 151: 280–288.
DOI:10.1016/j.chemosphere.2016.02.089
|
Rodriguez S, Vasquez L, Costa D, et al. 2014. Oxidation of Orange G by persulfate activated by Fe(Ⅱ), Fe(Ⅲ) and zero valent iron (ZVI)[J]. Chemosphere, 101(3): 86–92.
|
Waldemer R H, Tratnyek P G, Richard L J, et al. 2007. Oxidation of chlorinated ethenes by heat-activated persulfate:Kinetics and products[J]. Environmental Science & Technology, 41(3): 1010–1015.
|
Wang J L, Xu L J. 2012. Advanced oxidation processes for wastewater treatment:Formation of hydroxyl radical and application[J]. Critical Reviews in Environmental Science & Technology, 42(3): 251–325.
|
Wang J, Chu L. 2016. Irradiation treatment of pharmaceutical and personal care products (PPCPs) in water and wastewater:An overview[J]. Radiation Physics & Chemistry, 125: 56–64.
|
Wang S, Wang J. 2016. Carbamazepine degradation by gamma irradiation coupled to biological treatment[J]. Journal of Hazardous Materials, 321: 639–646.
|
王莹, 魏成耀, 黄天寅, 等. 2017. 氮掺杂碳纳米管活化过一硫酸盐降解酸性橙AO7[J]. 中国环境科学, 2017, 37(7): 2583–2590.
DOI:10.3969/j.issn.1000-6923.2017.07.021 |
Zhao Q, Mao Q, Zhou Y, et al. 2017. Metal-free carbon materials-catalyzed sulfate radical-based advanced oxidation processes:A review on heterogeneous catalysts and applications[J]. Chemosphere, 189: 224–238.
DOI:10.1016/j.chemosphere.2017.09.042
|
Zhou Y, Jiang J, Gao Y, et al. 2017. Activation of peroxymonosulfate by phenols:Important role of quinone intermediates and involvement of singlet oxygen[J]. Water Research, 125: 209–218.
DOI:10.1016/j.watres.2017.08.049
|
Zhou Y, Jiang J, Gao Y, et al. 2015. Activation of peroxymonosulfate by benzoquinone:A novel nonradical oxidation process[J]. Environmental Science & Technology, 49(21): 12941–12950.
|