 2018, Vol. 38
2018, Vol. 38



2. 复旦大学环境科学与工程系, 上海市大气颗粒物污染与防治重点实验室, 上海 200433
2. Shanghai Key Laboratory of Atmospheric Particle Pollution and Prevention, Department of Environmental Science and Engineering, Fudan University, Shanghai 200433
偶氮染料是工业常用染料, 其生产废水具有色度高、成分复杂、毒性强和难降解等特点(Almeida et al., 2014).因偶氮染料分子较为稳定, 并且具有较强的生物毒性, 因此难以被生物法处理.现代工业大多通过物化工艺, 例如吸附、膜分离、氧化等方式将其去除(Forgacs et al., 2004).其中, 高级氧化技术(AOPs)通过产生氧化能力强的活性自由基(如羟基自由基HO·), 不仅能够对偶氮染料进行脱色, 也具有较高的矿化率, 因此受到广泛关注(Narayani et al., 2017).此外, 污水中磷也广泛存在.过多的磷排放至水体会导致富营养化, 因此磷在污水排放中受到严格控制(Wilfert et al., 2015).传统的市政水处理包括1级、2级和3级处理过程, 但只有3级处理才能除磷(Zhao et al., 2016; Jin et al., 2014).然而, 当前许多地方, 尤其在中国农村, 还达不到3级处理的工艺要求.因此, 寻求高效低耗的除磷方法是目前除磷研究的热点之一.
近年来, 基于硫酸根自由基(SO4·-)的新型AOPs逐渐兴起并迅速发展.与HO·(E0=1.9~2.7 V)相比, SO4·-不但具有相似强氧化性(E0=2.5~3.1 V)(Buxton et al., 1988; Neta et al., 1988), 还拥有更长的半衰期和更好的选择性.说明SO4·-与目标污染物的可接触时间更长, 可能更利于降解目标污染物(Nie et al., 2014; 廖云燕等, 2014).过硫酸盐(PS)可以通过热、光、碱、过渡金属等方式活化分解得到SO4·-(Matzek and Carter, 2016; Nie et al., 2018; 段美娟等, 2017; 马京帅等, 2016).其中, Fe2+活化PS所需活化能低于其他活化方式(式(1)), 并且Fe2+具有成本低、毒性低等优点, 因此被广泛使用(Wang and Wang, 2018; Ji et al., 2014).然而, Fe2+在碱性条件下活化PS效果差.同时, Fe2+在与PS反应的初期即被迅速消耗, 使得反应瞬间终止.此外, 超量的Fe2+能够淬灭SO4·-(式(2))(Nie et al., 2015).研究表明, 零价铁(ZVI)活化PS的效果比Fe2+好.这是因为ZVI能够循环Fe3+生成Fe2+(式(3)), 从而减少铁沉淀的生成(Graça et al., 2017; Deng et al., 2014).同时, ZVI可以缓慢释放Fe2+, 从而使得活化PS的反应持续进行(式(4)~(5))(Wei et al., 2016; Li et al., 2015).此外, ZVI可以通过电子转移活化PS产生SO4·-(式(6))(Wei et al., 2016).因此, ZVI-PS体系已经成功的运用到多种难降解有机污染物的处理中(Zhu et al., 2016; Wu et al., 2018; Zhen et al., 2018).
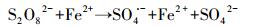
|
(1) |
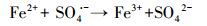
|
(2) |
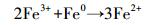
|
(3) |
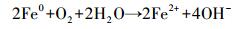
|
(4) |
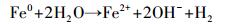
|
(5) |
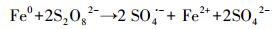
|
(6) |
此前的研究大多关注ZVI-PS体系对单一有机污染物的降解(Li et al., 2014; Li et al., 2015; Nie et al., 2015).在ZVI-PS体系中, 随着反应的进行, ZVI最终会转化为Fe3+.而Fe3+可以通过与磷酸盐反应形成难溶化合物沉淀(式(7)), 从而达到除磷的效果(嵇斌等, 2018).虽然有一些报道考虑到了磷酸盐对ZVI-PS体系降解有机污染物的影响(Nie et al., 2015), 然而, 应用ZVI-PS体系同时去除有机污染物和磷酸盐的研究极少, 只有Zhao等(2016)的研究表明该体系可以同时去除水中的双酚A和磷酸盐.但ZVI-PS体系能否成功地应用到其他类型的有机污染物(如染料)还未可知.此外, 目前对ZVI-PS体系的研究大多以纯水为主(李欢旋等, 2014; Deng et al., 2014; Wang et al., 2016).在实际污水中, 除了目标污染物, 还有多种类型的共存物质, 例如各种离子和溶解性有机质, 而这些共存物质有可能会抑制或促进体系的降解效果(Wang et al., 2016; Wei et al., 2016).有研究表明, 水中共存的Cl-、CO32-/HCO3-和NO2-与SO4·-和HO·的反应速率较快(Lutze et al., 2015; Li et al., 2017), 而腐植酸(HA)可能会覆盖在ZVI表面(Nie et al., 2015).因此, 考察ZVI-PS体系同步去除水中有机污染物和磷的同时, 还要考虑当其他物质存在时的影响.
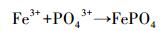
|
(7) |
酸性橙7(AO7)是目前全世界生产和使用量较高的偶氮染料(Harmer et al., 1992).为此, 本研究以AO7和磷酸盐为模型污染物, 将首次尝试研究ZVI-PS体系中AO7和磷的去除情况, 探讨各影响因素(如ZVI投加量、PS投加量、初始pH、多种无机离子和HA)对去除这两种污染物的影响, 并对比了不同水体(超纯水、自来水、河水、湖水和污水处理厂出水)中的去除效果, 以期为评价ZVI-PS体系同时去除水中AO7和磷的应用可行性提供依据.
2 材料与方法(Materials and methods) 2.1 实验材料AO7、磷酸二氢钾、PS、ZVI、硫酸、氢氧化钠、抗坏血酸、钼酸铵、酒石酸锑钾、乙醇(EtOH)和叔丁醇(TBA)均为分析纯购自国药集团化学试剂有限公司; HA为分析纯购自西亚试剂; 二氯甲烷(HPLC级)购于CNW公司.实验所有溶液均采用超纯水(电阻率18.2 MΩ·cm-1)配制.
2.2 降解实验ZVI活化PS同时去除水中AO7和磷的实验在500 mL烧杯中进行.加入一定浓度的AO7、磷酸二氢钾和PS的混合溶液后, 在磁力搅拌器上不断搅拌(200 r·min-1), 投加相应浓度的ZVI, 开始反应并计时, 在设定的时间点迅速取样.样品迅速加入过量的乙醇, 以清除溶液中残余的活性氧物种, 经0.22 μm的滤膜过滤后待测.除了考察溶液初始pH值的影响时, 需要用0.1 mol·L-1硫酸或氢氧化钠溶液调至特定pH值, 其他体系的pH值均不做任何调节, 并且在反应过程中也不做任何控制.
为了验证ZVI-PS在实际水体中的应用, 采集了4种不同的实际水体, 包括自来水、赣江河水、瑶湖湖水和某高校生活污水.水样带回实验室后使用滤膜(0.45 μm)进行过滤得到过滤液.分别用4种不同水样的过滤液配制0.05 mmol·L-1 AO7和5 mg·L-1磷, 用0.1 mol·L-1硫酸或氢氧化钠溶液调节pH至5.0后, 按照纯水中同样的操作流程进行ZVI-PS处理试验.
2.3 分析方法AO7的浓度利用UV-vis分光光度计(UV-5500, 元析)在其最大吸收波长484 nm处测量滤液的吸光度, 代入标准曲线求得其浓度.总有机碳(TOC)采用TOC测定仪(TOC-LCpH, 岛津)测定.磷残余浓度采用钼酸盐分光光度计法(GB-11893-89)测定.AO7溶液的UV-vis光谱利用UV-vis分光光度计(UV2600, 岛津)检测.Fe2+浓度采用邻菲罗啉法测定.测定总溶解性铁(Total Dissolved Iron, TDI)时利用盐酸羟胺还原Fe3+至Fe2+, 再利用邻菲罗啉测定总Fe2+浓度.TDI与Fe2+浓度的差值即为Fe3+的浓度.
AO7的降解产物采用GC-MS进行分析.取反应40 min时的反应液200 mL, 加入适量二氯甲烷(15 mL每次, 共3次)进行液-液萃取, 振荡15 min, 静置分层后吸取二氯甲烷萃取液, 蒸发浓缩至1 mL后, 通过GC-MS(Trace1300-ISQ, 赛默飞世尔)检测, 具体方法参照作者前期工作(聂明华等, 2018).通过对比NIST库推测产物结构.
2.4 反应动力学模型ZVI-PS体系降解有机污染物时, 污染物的降解规律一般符合准一级反应动力学模型, 因此本次研究采用该模型来考察不同环境下ZVI-PS系统对水中AO7的去除规律.模型如(式(8))所示.
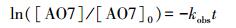
|
(8) |
式中, kobs表示AO7降解的准一级反应速率, [AO7]0为初始浓度, [AO7]为反应t时刻后的浓度, t为反应时间.
3 结果与讨论(Results and discussion) 3.1 ZVI-PS体系降解AO7 3.1.1 ZVI投加量的影响单独ZVI、单独PS和ZVI-PS 3种体系中AO7的降解情况如图 1a所示.当AO7的浓度为0.05 mmol·L-1, PS的投加量为1 mmol·L-1, ZVI投加量为0.75 mmol·L-1时, 在60 min内, 单独ZVI和单独PS体系中AO7基本没有降解, 而ZVI-PS体系中AO7的降解率达到了100%, 可见ZVI-PS体系可以有效去除水中AO7.在ZVI-PS体系中, 随着ZVI投加量从0.1 mmol·L-1增加到0.75 mmol·L-1, AO7的脱色率(60 min内)从32.35%增加到了100%(图 1b).这是因为ZVI的投加量越多, 产生活性氧化物种就越多(Nie et al., 2015).但当ZVI投加量大于0.75 mmol·L-1时, AO7的脱色率并没有继续提升, 反而会相应降低, 比如ZVI投加量分别是2 mmol·L-1和3 mmol·L-1时, AO7的脱色率降至93.29%和89.82%.这有可能是因为投入过多的ZVI时, 溶液中可能会产生过量的Fe2+, 从而淬灭了体系中的SO4·-, 降低了体系的效能(Bu et al., 2017).因此, 后续试验中ZVI的投加量选为0.75 mmol·L-1.
 |
| 图 1 不同体系对AO7去除率的影响(a)、初始ZVI的投加量对AO7在ZVI-PS体系中的降解影响(b)及初始PS的投加量影响(c) Fig. 1 Effects of different conditions on removal efficiency of AO7 (a), effect of initial ZVI dosage (b), effect of initial PS dosage(c) |
为考察不同PS投加量对ZVI/PS体系降解AO7的影响, 在AO7浓度为0.05 mmol·L-1, ZVI投加量为0.75 mmol·L-1, PS的投加量分别为0.1、0.5、0.75、1、1.5、1.75和2 mmol·L-1的条件下进行体系降解AO7的试验, 结果如图 1c所示.体系反应60 min时AO7的脱色率分别为5.68%、92.21%、100%、100%、100%、100%和100%.可见随着PS投加量的加大, AO7的脱色率也随之提升.这可能是因为PS投加量越高, 单位时间内产生的活性氧物种也更多, 因此体系的效率也越高(Wang et al., 2014).然而, 当前我国在《地表水环境质量标准(GB3838-2002)》和《生活饮用水卫生标准(GB5749-2006)》中, SO42-的浓度限值为250 mg·L-1 (2.6 mmol·L-1).因此, 虽然PS投加量越多, AO7的脱色率越高, 但并不能一味的提高PS的浓度.此外, 从图 1c可知, PS投加量超过0.75 mmol·L-1后, AO7的脱色率提升不明显.为此, 从保护环境和控制成本的角度上, 后续实验均选择0.75 mmol·L-1 PS和0.75 mmol·L-1 ZVI作为反应条件.
3.2 ZVI-PS体系同时去除AO7和磷 3.2.1 磷投加量的影响磷在城市和工业污水中广泛存在, 并被认为是导致湖泊富营养化的主要因素之一.考察了AO7浓度为0.05 mmol·L-1, ZVI和PS投加量均为0.75 mmol·L-1, 磷的投加量分别为2、5、10和15 mg·L-1时对ZVI-PS体系同时去除AO7和磷的影响(图 2).如图 2所示, 在ZVI-PS体系中, 低浓度的磷(2 mg·L-1)对AO7的降解基本没有影响, 但较高浓度的磷(5~15 mg·L-1)有明显的抑制作用.然而, 反应60 min后, 磷的投加量分别为2、5、10和15 mg·L-1时, AO7的脱色率也能达到98.88%、96.95%、92.19%和84.77%.同样地, 有研究表明, 磷会抑制ZVI-PS体系降解水中双酚A(Zhao et al., 2016)、苯达松(Wei et al., 2016)和氯霉素(Nie et al., 2015).在ZVI-PS体系中, 主要的活性氧物种是HO·和SO4·-.而HO·和SO4·-都是具有强氧化能力的自由基, 可以与水溶液中的H2PO4-发生反应, 将H2PO4-/HPO42-转变成H2PO4·(Maruthamuthu and Neta, 1978).值得注意的是, H2PO4·在不同的pH条件下可以转化成HPO4·或PO4·2-(式(9)~(11)), 但这3种磷酸根自由基的氧化能力均弱于HO·和SO4·-(Maruthamuthu and Neta, 1978; Rosso et al., 1998).本研究中, ZVI-PS体系的初始pH值均 < 5.7, 因此, H2PO4·是体系中主要的活性粒子.H2PO4-浓度较低时(2 mg·L-1), 只会消耗少量的HO·或SO4·-, 而生成的H2PO4·也可以参与AO7的降解, 因此对ZVI-PS体系的降解效果影响不大.然而, 当H2PO4-浓度超过2 mmol·L-1时, AO7的降解开始受到抑制, 这可能是因为过多的HO·或SO4·-被H2PO4-淬灭而生成活性较弱的H2PO4·.
 |
| 图 2 初始磷的投加量对ZVI-PS同时去除AO7和磷效果的影响 Fig. 2 Effects of initial P dosage on AO7 and P removal in ZVI-PS system |
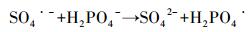
|
(9) |
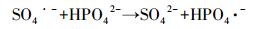
|
(10) |
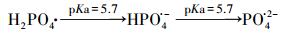
|
(11) |
通过检测ZVI-PS体系中Fe2+的浓度变化(图 3a), 表明ZVI在PS存在的情况下被腐蚀, 能够持续不间断地释放Fe2+(0.20~12.74 mg·L-1), 并且ZVI的存在还可能会促进Fe3+向Fe2+的转化.生成的Fe2+再与PS反应生成Fe3+和强氧化性的自由基, 从而形成Fe2+和Fe3+的循环, 保证了ZVI-PS体系中AO7较高的脱色速率.ZVI-PS体系中, ZVI慢慢被PS腐蚀, 并在过量PS的存在情况下可以最终转化成Fe3+(2.80~22.16 mg·L-1).而Fe3+可以与磷反应生成难溶性盐与水体分离, 因此在污水处理中常被用来除磷(嵇斌等, 2018).基于此, 猜测ZVI-PS体系可能可以同时去除AO7和磷.
 |
| 图 3 ZVI-PS体系(a)和ZVI-PS-P体系(b)中Fe2+、TDI和Fe3+值随时间的变化曲线 Fig. 3 Time course of Fe2+, TDI and Fe3+ during AO7 degradation reaction in (a) ZVI-PS and (b) ZVI-PS-P system |
磷的添加对反应系统中的Fe2+及TDI含量具有显著影响.无磷存在时, ZVI的分解较快, 并生成较多的Fe2+, (图 3a).而相比之下, 有磷存在时, 溶液中的Fe2+较少(图 3b).同样的, 磷的存在也使得体系中TDI的含量明显降低, 这可能是因为体系中生成的Fe3+与磷反应生成沉淀, 阻碍了Fe3+与ZVI之间的反应, 进一步使得体系中Fe2+含量较低.为了验证ZVI-PS体系同时去除AO7和磷的能力, 检测了反应60 min后溶液中磷的残留量.由图 2插图可知, 磷的投加量分别为2、5、10和15 mg·L-1时, 磷的去除率能达到100%、100%、100%和87.51%, 说明ZVI-PS体系在降解AO7的同时, 还可以有效去除浓度范围较广的磷, 而这个具有重要经济价值的潜在功能在以往研究中往往被忽视.
3.2.2 pH值的影响溶液pH值是影响ZVI-PS体系去除AO7和磷的重要因素之一.在磷的初始浓度为5 mg·L-1时, 考察了不同初始pH条件下ZVI-PS体系中AO7和磷的去除效果.结果显示(图 4a), ZVI-PS体系氧化降解AO7的效率随着反应体系初始pH升高而降低.在初始pH分别为3、5、5.5、6、6.5、7、9和11的条件下, 反应60 min后, AO7的降解率分别为95.45%、91.71%、58.83%、38.08%、35.11%、1.40%、0.55%和0.11%.这是因为酸性pH值条件有利于ZVI的分解, 并生成溶解态的Fe2+, 从而进一步激发产生AO7降解所需的SO4·-.而相比之下, 中性或碱性条件下, 磷酸盐的缓冲作用使得体系pH降低速率减慢(图 4b), 不利于ZVI的分解, 因此, AO7的去除率迅速降低.Wang等(2014)的研究发现, 碱性环境下, ZVI-PS体系中释放的Fe2+浓度低于0.01 mmol·L-1.
 |
| 图 4 溶液初始pH对ZVI-PS同时去除AO7和磷效果的影响(a)及pH值随时间的变化曲线(b) Fig. 4 Effects of initial P dosage on AO7 and P removal in ZVI-PS system(a) and evolution of pH values(b) |
从图 4a插图可以看出, 初始pH分别为3、5、5.5、6、6.5、7、9和11的条件下, 磷的去除率分别为95.73%、100%、90.12%、81.23%、52.31%、13.17%、5.32%和3.25%, 说明中性和碱性环境下不利于磷的去除, 这可能是因为ZVI在中性和碱性条件下的腐蚀速率不高, 生成的Fe3+不多.值得注意的是, 磷在pH = 3时的去除率小于pH = 5时, 这可能是由于Fe3+在pH > 4的条件下会发生水解形成羟基络合物(Nie et al., 2018), 而这些络合物可以通过吸附、电中和等形式去除水中的磷, 因此在除磷的过程中起到了一定的辅助作用(刘宁等, 2012).总的来说, ZVI-PS体系在中性和碱性条件下去除AO7和磷的效果均不好, 因此在实际应用时, 应适当降低碱度, 以确保体系效果.
3.2.3 自由基淬灭实验一般来说, 在活化PS的体系中, 通常会产生SO4·-和HO·.在ZVI和PS投加量均为0.75 mmol·L-1, 磷的投加量为5 mg·L-1时, 通过投加EtOH和TBA, 来验证ZVI-PS体系中主要的活性氧物种(SO4·-或HO·).EtOH与SO4·-和HO·反应速率常数相近并且都较快, 分别为kHO· = 1.2× 109~2.8 × 109 L·mol-1·s-1; kSO4·-=1.6× 107~7.7×107 L·mol-1·s-1, 因此可以作为SO4·-和HO·的淬灭剂.TBA与HO·的反应速率常数较大(kHO·=3.8×108~7.6×108 L·mol-1·s-1), 而与SO4·-的反应速率常数较小(kSO4·-=4×105~9.1 × 105 L·mol-1·s-1), 因此只是HO·的淬灭剂(Anipsitakis et al., 2004).通过比较投加EtOH和TBA后的去除效果, 可以确定体系中的主要活性氧物种.由图 5可知, EtOH和TBA加入ZVI-PS体系后, 均会不同程度的降低体系的降解效果.比如, 引入EtOH后, AO7的去除率较低, 并且随着EtOH投加量的增加, AO7的去除率不断降低.加入7.5和75 mmol·L-1 EtOH后, 60 min时AO7的降解率分别降至70.28%和38.91%, 说明体系中SO4·-或HO·参与了AO7的氧化降解.而加入7.5和75 mmol·L-1 TBA后, 60 min时AO7的降解率仍然可以达到86.40%和82.80%, 说明EtOH的抑制效果明显强于TBA.因此, 虽然SO4·-和HO·对AO7的降解均有一定的贡献, 但相较而言, SO4·-起到了主要作用.
 |
| 图 5 ZVI-PS-P体系中自由基淬灭剂对AO7降解的影响 Fig. 5 Effects of AO7 degradation on ZVI-PS-P system in the presence of different radical scavengers |
天然水体中广泛存在着各种阴离子和溶解性有机质(DOM), 研究发现它们会对SO4·-降解有机污染物产生或多或少的影响(Li et al., 2015).实验考察了水中常见阴离子(Cl-、HCO3-/CO32-、NO3-和NO2-)和腐植酸(HA)存在时([AO7]0 = 0.05 mmol·L-1, [PS]0 = 0.75 mmol·L-1, [ZVI]0 = 0.75 mmol·L-1, [P]0 = 5 mg·L-1, [阴离子]0 = 0.01~5 mmol·L-1, [HA]0 = 2~8 mg·L-1), ZVI-PS体系去除AO7和磷的情况.AO7降解的准一级反应速率(kobs)如表 1所示, 可以看出, ZVI-PS体系中添加阴离子和HA会影响AO7的降解速率, 并且不同物质的作用差异较大.
| 表 1 共存物质对ZVI-PS-P体系同时去除水中AO7和磷的影响 Table 1 Simultaneous removal of AO7 and phosphate by ZVI-PS-P system in the presence of various substances |
Cl-会轻微抑制AO7的降解, 并且Cl-浓度越高, 降解速率越慢.有研究表明, Cl-可与SO4·-和HO·发生反应, 如SO4·-可以将Cl-转化成Cl·(k = 3.2×108 L·mol-1·s-1), HO·也可以与Cl-反应生成ClHO·-(k = 4.3×109 L·mol-1·s-1)和Cl·(k = 4.3×1010 L·mol-1·s-1)(Buxton et al., 1988; Neta et al., 1988).虽然这些含氯自由基也可能会参与AO7的降解, 但它们的氧化能力比SO4·-和HO·更弱.并且, 随着Cl-浓度的升高, 体系中更多的SO4·-或HO·会被Cl-消耗(Fang et al., 2017).因此, AO7的降解效率随之降低.虽然Cl-会抑制AO7的降解, 但磷的去除率依然可以达到100%, 这可能是因为ZVI在Cl-的存在下腐蚀不受影响, 甚至更快, 因此可能产生更多的Fe3+.
实验同时发现, 即使在较低浓度下(如0.1 mmol·L-1), 投加的NO2-对AO7的降解起显著抑制作用.这是因为NO2-对SO4·-及HO·自由基具有较强的淬灭作用.加入NO2-后, 磷的去除率也有一定程度的降低.比如, 投加5 mmol·L-1NO2-后, 磷的去除率降至94.62%.这可能是因为AO7在ZVI-PS体系的降解过程中会生成带有羧酸官能团的中间产物, 从而降低体系pH(Li et al., 2014), 加速ZVI的腐蚀.然而NO2-的存在使得AO7的降解受阻, 使得体系pH维持在较高的水平(表 1), 从而导致磷的去除率降低.
与Cl-相似, NO3-也会轻微抑制AO7的降解, 并且随NO3-浓度的增加AO7的降解速率降低.这可能是因为NO3-会与AO7竞争SO4·-或HO·, 反应生成了氧化能力更弱的NO3·, 从而削弱了ZVI-PS体系的氧化能力.此外, 有研究表明, 通过夺取ZVI表面的电子, NO3-能与ZVI反应生成NO2-及NH3(Zhang et al., 2017).因此, 相比于无NO3-的水溶液, Fe2+在含NO3-的环境中更易被氧化成Fe3+.而且, NO3-与ZVI的反应是一个腐蚀过程, 容易在ZVI表面生成膜状物而阻碍PS活化反应的进一步进行(Su et al., 2012).虽然NO3-会轻微抑制AO7的降解, 但磷的去除率也可以达到100%.
HCO3-能够抑制ZVI-PS体系降解AO7, 并且抑制作用随着HCO3-浓度的升高而明显加强.投加0.01 mmol·L-1 HCO3-后, AO7在60 min内的去除率从96.95%降低至96.56%.而投加0.1和1 mmol·L-1 HCO3-后, AO7去除率则分别大幅度降低至42.53%和2.28%.体系中加入HCO3-后, 溶液的pH值会发生改变(pH = 4.44~7.18)(表 1).通过3.2.2节的讨论可知, AO7在中性和碱性条件下的降解速率明显低于酸性条件, 因此, HCO3-可能主要通过改变体系的pH值来抑制AO7降解速率.在较高的pH环境下, 水溶液中的Fe3+和Fe2+会迅速生成沉淀, 从而影响SO4·-的产生.此外, HCO3-和CO32-都可以强烈地与AO7竞争体系中的活性粒子, 比如SO4·-和HO·.虽然, HCO3-和CO32-可以与SO4·-和HO·生成活性较低的无机自由基, 如CO3·-和HCO3·, 但这些新生成的自由基的氧化能力均无法与SO4·-和HO·媲美, 从而降低了体系的降解效率(Nie et al., 2014; Li et al., 2017).而且, 随着pH的升高, SO4·-和HO·的生成量迅速降低, 这样使得CO3·-和HCO3·的生成量也随之减少.此外, HCO3-能够抑制ZVI-PS体系去除磷, 并且抑制作用随着HCO3-浓度的升高而明显加强.
HA对AO7的降解产生了一定的抑制作用, 并且随着投加量的升高, 抑制程度也随之略微增加.当加入的HA浓度为2 mg·L-1时, AO7的降解率降低到89.29%.继续增加HA对体系的影响并不明显, 如投加4和8 mg·L-1 HA后, AO7的去除率与投加2 mg·L-1 HA的情况相似, 分别为88.43%和88.20%.HA的分子中具有多种活性基团, 比如羟基、羧基、胺基和酚羟基, 这些基团可以与SO4·-和HO·进行反应(Liu et al., 2018).因此, 体系中存在的HA会和AO7竞争SO4·-或HO·, 导致AO7的降解受到抑制.然而, SO4·-与溶解性有机质(DOM)之间的反应速率较慢(Luo et al., 2017), 因此继续添加HA对体系的影响不明显.
3.2.5 实际水体中AO7和磷的去除为了研究ZVI-PS体系在实际水体中同时去除AO7和磷的效果, 选取了4种不同来源的实际水体进行试验:自来水、赣江河水、鄱阳湖湖水、某高校生活污水.4种水样的水质参数及AO7在其中的反应速率常数如表 2所示.实验结果表明, AO7在不同水体中降解的速率为:超纯水>自来水>河水>湖水>生活污水.这可能是因为实际水样中成分复杂, 含有较多的溶解性有机质以及各种离子, 会与AO7竞争SO4·-和HO·, 因此可能会影响到AO7的去除效果.并且从表 2中可以看出, 水样中溶解性有机碳(DOC)含量越高, AO7的降解速率越低.但是, 值得注意的是, 尽管实际水样中AO7的降解速率会变慢, 反应80 min后, AO7和磷的去除率仍然可分别达到80.31%~92.69%和87.54%~100%.因此, ZVI-PS可以有效地同时去除实际水体中的AO7和磷.
| 表 2 不同水体的理化性质参数以及AO7在其中的反应速率常数 Table 2 Physical and chemical property parameters of different waters and AO7 in which the reaction rate constant |
AO7的初始浓度为0.05 mmol·L-1, 磷的浓度为5 mg·L-1, ZVI和PS的投加量均为0.75 mmol·L-1, 在反应过程中, 随着反应时间的延长, 溶液中的TOC值逐渐降低.如图 6所示, 反应60 min后, TOC的去除率为30.11%.虽然AO7的去除率可以达到96.95%, 并且实现了一定程度的矿化, 但是矿化程度不高, 说明体系中还存在着大量难以矿化的有机中间产物.
 |
| 图 6 AO7在ZVI-PS-P体系降解过程中TOC的变化 Fig. 6 Degradation efficiency of AO7 and removal rate of TOC in ZVI-PS-P system |
图 7为ZVI-PS体系降解AO7过程中UV-vis光谱随反应时间的变化.从图 7中可以看到, AO7有4个特征峰.位于484 nm和430 nm的特征峰主要是因为发色基团偶氮键, 而位于310 nm和230 nm处的特征峰分别是因为萘环和苯环结构(Wang et al., 2004).随着反应时间的延长, 位于可见光波段的吸收峰迅速下降直至消失, 说明发色基团不断被破坏.反应5 min后, 位于紫外波段的区域形成新的吸收峰, 说明有新的中间产物形成, 并且这些中间产物可能还含有苯环或萘环(Zubir et al., 2014).值得注意的是, 这些新形成的吸收峰在随后的反应中逐渐下降, 表明这些中间产物可以继续被氧化降解.反应后期, 随着ZVI不断被腐蚀, 溶液中的Fe3+不断增加, 因此使得紫外吸收波段吸收峰不断升高.
 |
| 图 7 AO7在ZVI-PS-P体系降解过程中紫外可见光谱的变化 Fig. 7 UV-vis spectra changes of AO7 solution treated with ZVI-PS-P system |
利用GC-MS对ZVI-PS体系去除AO7和磷过程中产生的产物进行了分析鉴定.AO7的颜色主要是因为其分子结构中存在偶氮键, 因此根据AO7的降解中间产物及其脱色规律, 可以推测出AO7在ZVI-PS体系中可能的降解路径(图 8).首先, ZVI-PS体系中生成的SO4·-或HO·可能首先会攻击AO7的偶氮键, 使其断键而迅速脱色, 同时形成了1-氨基-2-萘酚和4-氨基苯磺酸(图 8).然而, GC-MS并没有检测到这两种化合物.本实验体系是开放体系, 并且在磁力搅拌的作用下不断搅拌.随着反应的进行, 溶解氧的浓度基本持平(8.3~8.4 mg·L-1).1-氨基-2-萘酚是一种对氧气较为敏感的物质, 容易在有氧条件下氧化(Zhao et al., 2010; Thung et al., 2018; Cai et al., 2016; Kudlich et al., 1999).本实验体系溶解氧浓度基本不变, 因此足够氧化1-氨基-2-萘酚.而4-氨基苯磺酸的熔点较高, 在当前进样口设定的温度下不能被气化(Qi et al., 2016).1-氨基-2-萘酚在体系内可能被氧化成邻苯二甲酰亚胺(产物1)和邻苯二甲酸(产物2), 而4-氨基苯磺酸继续被氧化形成对苯醌(产物3)(图 8).这些中间产物降解成其他小分子酸的有机化合物, 最终矿化成CO2、H2O和无机盐.
 |
| 图 8 ZVI-PS-P体系中AO7的可能降解路径 Fig. 8 Possible degradation pathways of AO7 in ZVI-PS-P system |
1) ZVI-PS体系可以有效地去除水中AO7, 体系中加入磷后会影响AO7的降解率.
2) ZVI-PS体系可以同时去除水中AO7和磷; 酸性条件下的AO7和磷的去除率较高; 淬灭实验结果表明, SO4·-和HO·是ZVI-PS体系中的主要活性物种, 并且SO4·-对AO7的降解起主要作用.
3) 水中共存的阴离子和HA对AO7和磷的去除均有抑制作用, 其对AO7和磷去除的抑制能力排序均为: HCO3- > NO2- > HA > NO3-> Cl-.
4) AO7和磷在不同水体中的去除率为:超纯水>自来水>河水>湖>生活污水.
5) AO7降解过程中偶氮键断裂, 产生的中间产物被进一步氧化, 形成其他物质.同时, AO7降解过程中TOC有所降低, 说明部分AO7被矿化为CO2和H2O.
Almeida E J R, Corso C R. 2014. Comparative study of toxicity of azo dye Procion Red MX-5B following biosorption and biodegradation treatments with the fungi Aspergillus niger and Aspergillus terreus[J]. Chemosphere, 112: 317–322.
DOI:10.1016/j.chemosphere.2014.04.060
|
Anipsitakis G P, Dionysiou D D. 2004. Radical generation by the interaction of transition metals with common oxidants[J]. Environmental Science & Technology, 38(13): 3705–3712.
|
Bu L, Bi C, Shi Z, et al. 2017. Significant enhancement on ferrous/persulfate oxidation with epigallocatechin-3-gallate:Simultaneous chelating and reducing[J]. Chemical Engineering Journal, 321: 642–650.
DOI:10.1016/j.cej.2017.04.001
|
Buxton G V, Greenstock C L, Helman W P, et al. 1988. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/O-) in aqueous solution[J]. Journal of Physical and Chemical Reference Data, 17(2): 513–886.
DOI:10.1063/1.555805
|
Cai C, Zhang Z, Liu J, et al. 2016. Visible light-assisted heterogeneous Fenton with ZnFe2O4 for the degradation of Orange Ⅱ in water[J]. Applied Catalysis B:Environmental, 182: 456–468.
DOI:10.1016/j.apcatb.2015.09.056
|
Deng J, Shao Y, Gao N, et al. 2014. Zero-valent iron/persulfate (Fe0/PS) oxidation acetaminophen in water[J]. International Journal of Environmental Science and Technology, 11(4): 881–890.
DOI:10.1007/s13762-013-0284-2
|
段美娟, 马邕文, 万金泉, 等. 2017. 应用铁铜双金属有机骨架活化过硫酸盐催化剂去除水中有机污染物的研究[J]. 环境科学学报, 2017, 37(5): 1742–1750.
|
Fang C, Lou X, Huang Y, et al. 2017. Monochlorophenols degradation by UV/persulfate is immune to the presence of chloride:Illusion or reality?[J]. Chemical Engineering Journal, 323: 124–133.
DOI:10.1016/j.cej.2017.04.094
|
Forgacs E, Cserhati T, Oros G. 2004. Removal of synthetic dyes from wastewaters:a review[J]. Environment international, 30(7): 953–971.
DOI:10.1016/j.envint.2004.02.001
|
Graça C A L, Fugita L T N, de Velosa A C, et al. 2017. Amicarbazone degradation promoted by ZVI-activated persulfate:study of relevant variables for practical application[J]. Environmental Science and Pollution Research, 25(2): 1–10.
|
Harmer C, Bishop P. 1992. Transformation of azo dye AO-7 by wastewater biofilms[J]. Water Science and Technology, 26(3/4): 627–636.
|
Ji Y, Ferronato C, Salvador A, et al. 2014. Degradation of ciprofloxacin and sulfamethoxazole by ferrous-activated persulfate:implications for remediation of groundwater contaminated by antibiotics[J]. Science of the Total Environment, 472: 800–808.
DOI:10.1016/j.scitotenv.2013.11.008
|
Jin L, Zhang G, Tian H. 2014. Current state of sewage treatment in China[J]. Water Research, 66: 85–98.
DOI:10.1016/j.watres.2014.08.014
|
嵇斌, 李杰, 豆宁龙. 2018. 不同价态铁强化SBR除磷研究[J]. 工业水处理, 2018, 38(3): 17–20.
|
Kudlich M, Hetheridge M J, Knackmuss H J, et al. 1999. Autoxidation reactions of different aromatic o-aminohydroxynaphthalenes that are formed during the anaerobic reduction of sulfonated azo dyes[J]. Environmental Science & Technology, 33(6): 896–901.
|
Kusic H, Peternel I, Ukic S, et al. 2011. Modeling of iron activated persulfate oxidation treating reactive azo dye in water matrix[J]. Chemical Engineering Journal, 172(1): 109–121.
DOI:10.1016/j.cej.2011.05.076
|
Li H, Wan J, Ma Y, et al. 2014. Influence of particle size of zero-valent iron and dissolved silica on the reactivity of activated persulfate for degradation of acid orange 7[J]. Chemical Engineering Journal, 237: 487–496.
DOI:10.1016/j.cej.2013.10.035
|
Li H, Wan J, Ma Y, et al. 2015. Role of inorganic ions and dissolved natural organic matters on persulfate oxidation of acid orange 7 with zero-valent iron[J]. RSC Advances, 5(121): 99935–99943.
DOI:10.1039/C5RA16094D
|
Li R, Jin X, Megharaj M, et al. 2015. Heterogeneous Fenton oxidation of 2, 4-dichlorophenol using iron-based nanoparticles and persulfate system[J]. Chemical Engineering Journal, 264: 587–594.
DOI:10.1016/j.cej.2014.11.128
|
Li W, Orozco R, Camargos N, et al. 2017. Mechanisms on the impacts of alkalinity, pH, and chloride on persulfate-based groundwater remediation[J]. Environmental Science & Technology, 51(7): 3948–3959.
|
李欢旋, 万金泉, 马邕文, 等. 2014. 不同粒径零价铁活化过硫酸钠氧化降解酸性橙7的影响及动力学研究[J]. 环境科学, 2014, 35(9): 3422–3429.
|
廖云燕, 刘国强, 赵力, 等. 2014. 利用热活化过硫酸盐技术去除阿特拉津[J]. 环境科学学报, 2014, 34(4): 931–937.
|
刘宁, 陈小光, 崔彦召, 等. 2012. 化学除磷工艺研究进展[J]. 化工进展, 2012, 31(7): 1597–1603.
|
Liu Y, Yang Y, Pang S, et al. 2018. Mechanistic insight into suppression of bromate formation by dissolved organic matters in sulfate radical-based advanced oxidation processes[J]. Chemical Engineering Journal, 333: 200–205.
DOI:10.1016/j.cej.2017.09.159
|
Luo S, Wei Z, Dionysiou D D, et al. 2017. Mechanistic insight into reactivity of sulfate radical with aromatic contaminants through single-electron transfer pathway[J]. Chemical Engineering Journal, 327: 1056–1065.
DOI:10.1016/j.cej.2017.06.179
|
Lutze H V, Kerlin N, Schmidt T C. 2015. Sulfate radical-based water treatment in presence of chloride:formation of chlorate, inter-conversion of sulfate radicals into hydroxyl radicals and influence of bicarbonate[J]. Water Research, 72: 349–360.
DOI:10.1016/j.watres.2014.10.006
|
Maruthamuthu P, Neta P. 1978. Phosphate radicals. Spectra, acid-base equilibriums, and reactions with inorganic compounds[J]. The Journal of Physical Chemistry, 82(6): 710–713.
DOI:10.1021/j100495a019
|
Matzek L W, Carter K E. 2016. Activated persulfate for organic chemical degradation:a review[J]. Chemosphere, 151: 178–188.
DOI:10.1016/j.chemosphere.2016.02.055
|
马京帅, 吕文英, 刘国光, 等. 2016. 吉非罗齐在热活化过硫酸盐体系中的降解机制研究[J]. 环境科学学报, 2016, 36(10): 3774–3783.
|
聂明华, 徐文丽, 张文静, 等. 2018. Fe2+和零价铁活化过一硫酸盐降解酸性橙7[J]. 环境科学学报, 2018, 38(10): 3997–4005.
|
Narayani H, Augustine R, Sumi S, et al. 2017. Removal of basic and industrial azo reactive dyes from aqueous solutions via Fenton-like reactions using catalytic non-magnetic Pd-flyash and magnetic Pd-Fe3O4-flyash composite particles[J]. Separation and Purification Technology, 172: 338–349.
DOI:10.1016/j.seppur.2016.08.027
|
Neta P, Huie R E, Ross A B. 1988. Rate constants for reactions of inorganic radicals in aqueous solution[J]. Journal of Physical and Chemical Reference Data, 17(3): 1027–1284.
DOI:10.1063/1.555808
|
Nie M, Yan C, Li M, et al. 2015. Degradation of chloramphenicol by persulfate activated by Fe2+ and zerovalent iron[J]. Chemical Engineering Journal, 279: 507–515.
DOI:10.1016/j.cej.2015.05.055
|
Nie M, Yan C, Xiong X, et al. 2018. Degradation of chloramphenicol using a combination system of simulated solar light, Fe2+ and persulfate[J]. Chemical Engineering Journal, 348: 455–463.
|
Nie M, Yang Y, Zhang Z, et al. 2014. Degradation of chloramphenicol by thermally activated persulfate in aqueous solution[J]. Chemical Engineering Journal, 246: 373–382.
DOI:10.1016/j.cej.2014.02.047
|
Qi C, Liu X, Ma J, et al. 2016. Activation of peroxymonosulfate by base:implications for the degradation of organic pollutants[J]. Chemosphere, 151: 280–288.
DOI:10.1016/j.chemosphere.2016.02.089
|
Rosso J A, Nieto F J R, Gonzalez M C, et al. 1998. Reactions of phosphate radicals with substituted benzenes[J]. Journal of Photochemistry and Photobiology A:Chemistry, 116(1): 21–25.
DOI:10.1016/S1010-6030(98)00266-4
|
Thung W E, Ong S A, Ho L N, et al. 2018. Biodegradation of Acid Orange 7 in a combined anaerobic-aerobic up-flow membrane-less microbial fuel cell:Mechanism of biodegradation and electron transfer[J]. Chemical Engineering Journal, 336: 397–405.
DOI:10.1016/j.cej.2017.12.028
|
Wang K, Zhang J, Lou L, et al. 2004. UV or visible light induced photodegradation of AO7 on TiO2 particles:the influence of inorganic anions[J]. Journal of Photochemistry and Photobiology A:Chemistry, 165(1/3): 201–207.
|
Wang Q, Shao Y, Gao N, et al. 2016. Degradation of alachlor with zero-valent iron activating persulfate oxidation[J]. Journal of the Taiwan Institute of Chemical Engineers, 63: 379–385.
DOI:10.1016/j.jtice.2016.03.038
|
Wang S, Wang J. 2018. Trimethoprim degradation by Fenton and Fe (Ⅱ)-activated persulfate processes[J]. Chemosphere, 191: 97–105.
DOI:10.1016/j.chemosphere.2017.10.040
|
Wang X, Wang L, Li J, et al. 2014. Degradation of Acid Orange 7 by persulfate activated with zero valent iron in the presence of ultrasonic irradiation[J]. Separation and Purification Technology, 122: 41–46.
DOI:10.1016/j.seppur.2013.10.037
|
Wei X, Gao N, Li C, et al. 2016. Zero-valent iron (ZVI) activation of persulfate (PS) for oxidation of bentazon in water[J]. Chemical Engineering Journal, 285: 660–670.
DOI:10.1016/j.cej.2015.08.120
|
Wilfert P, Kumar P S, Korving L, et al. 2015. The relevance of phosphorus and iron chemistry to the recovery of phosphorus from wastewater:a review[J]. Environmental Science & Technology, 49(16): 9400–9414.
|
Wu S, He H, Li X, et al. 2018. Insights into atrazine degradation by persulfate activation using composite of nanoscale zero-valent iron and graphene:Performances and mechanisms[J]. Chemical Engineering Journal, 341: 126–136.
DOI:10.1016/j.cej.2018.01.136
|
Zhang Y, Douglas G B, Pu L, et al. 2017. Zero-valent iron-facilitated reduction of nitrate:Chemical kinetics and reaction pathways[J]. Science of the Total Environment, 598: 1140–1150.
DOI:10.1016/j.scitotenv.2017.04.071
|
Zhao H Z, Sun Y, Xu L N, et al. 2010. Removal of Acid Orange 7 in simulated wastewater using a three-dimensional electrode reactor:Removal mechanisms and dye degradation pathway[J]. Chemosphere, 78(1): 46–51.
DOI:10.1016/j.chemosphere.2009.10.034
|
Zhao L, Ji Y, Kong D, et al. 2016. Simultaneous removal of bisphenol A and phosphate in zero-valent iron activated persulfate oxidation process[J]. Chemical Engineering Journal, 303: 458–466.
DOI:10.1016/j.cej.2016.06.016
|
Zhen G, Lu X, Su L, et al. 2018. Unraveling the catalyzing behaviors of different iron species (Fe2+ vs. Fe0) in activating persulfate-based oxidation process with implications to waste activated sludge dewaterability[J]. Water Research, 134: 101–114.
DOI:10.1016/j.watres.2018.01.072
|
Zhu C, Fang G, Dionysiou D D, et al. 2016. Efficient transformation of DDTs with persulfate activation by zero-valent iron nanoparticles:a mechanistic study[J]. Journal of Hazardous Materials, 316: 232–241.
DOI:10.1016/j.jhazmat.2016.05.040
|
Zubir N A, Yacou C, Zhang X, et al. 2014. Optimisation of graphene oxide-iron oxide nanocomposite in heterogeneous Fenton-like oxidation of Acid Orange 7[J]. Journal of Environmental Chemical Engineering, 2(3): 1881–1888.
DOI:10.1016/j.jece.2014.08.001
|