 2018, Vol. 38
2018, Vol. 38



2. 南京农业大学资源与环境科学学院, 南京 210095
2. College of Resources and Environmental Sciences, Nanjing Agricultural University, Nanjing 210095
酸性矿山废水(AMD)不仅酸度高, 同时含有大量Fe、硫酸根及重(类)金属元素(Wei et al., 2013;Vhahangwele, 2016;Wu et al., 2016).此类废水不仅会腐蚀井下管道及排水泵等设备, 亦将严重污染地表水及土地资源, 因而引起国内外学者的广泛关注(Liu et al., 2013).目前, AMD主要采用石灰中和法治理(Song et al., 2014;Meschke et al., 2015;Lee et al., 2016).但因石灰需要量较大导致处理成本高, 且中和产物Fe(OH)3、Fe(OH)2、CaSO4等易黏附在石灰表面而影响中和效率, 并导致大量废渣产生, 带来二次污染风险.因此, 探索Fe、硫酸根及重(类)金属元素的安全、环保去除对治理AMD具有重要意义.
嗜酸性氧化亚铁硫杆菌(A.ferrooxidans)能够高效催化酸性硫酸盐环境中Fe2+向Fe3+转化, 并伴随着Fe3+水解产生施氏矿物、黄铁矾等次生铁矿物(宋永伟等, 2013;Zhu et al., 2013).具体反应式如下:
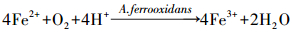
|
(1) |
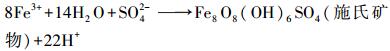
|
(2) |
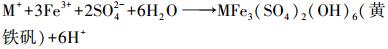
|
(3) |
式中,M+为K+、NH4+、Na+等一价阳离子.
施氏矿物具有纳米级粒度和不规则孔道结构, 比表面积多在100~200 m2·g-1, 且富含羟基、硫酸根等基团(Jönsson et al., 2005;Waychunas et al., 2005;Bang et al., 2005).黄铁矾具有不溶于稀酸, 易于沉淀、洗涤和过滤等优点, 也是性能优异、稀有昂贵的赭黄色无机颜料(周顺桂等, 2004), 且在作为固定化载体时有助于A.ferrooxidans生物膜的形成(Karamanev, 1991;Elgersma et al., 1993;Dutrizac, 1996;Gómez et al., 2000).已有的研究结果证实, 施氏矿物和黄铁矾对重(类)金属离子均有较大的吸附及共沉淀作用, 是较为理想的吸附材料(Gan et al., 2015;Mihone et al., 2015;Zhang et al., 2016), 对重金属废水的治理具有潜在应用价值.由上启示, 生物成因施氏矿物和黄铁矾不仅可以有效去除Fe、硫酸根, 还可通过吸附或共沉淀方式清除有毒元素.促使酸性硫酸盐环境中Fe向次生铁矿物转变具有一定的科学研究价值.
影响次生铁矿物形成的因素很多, 其中最主要的是溶液的酸度、反应时间、反应温度、能源物质Fe2+浓度、一价阳离子种类/浓度、晶种种类等(Dutrizac et al., 1976;Dutrizac, 1999;邓志明等, 2002).根据化学方程式(1)~(3)可知, 施氏矿物和黄铁矾生物合成的实质是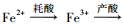
鉴于此, 本研究以去除AMD中Fe、硫酸根及重(类)金属元素为目的, 通过A.ferrooxidans介导的生物矿化法, 研究不同AMD环境下Na+对体系Fe2+氧化率、总Fe沉淀率、次生铁矿物矿相等的影响.以期为促使酸性硫酸盐环境中Fe向次生铁矿物的转变和调控提供必要的理论依据.
2 材料与方法(Materials and methods) 2.1 供试材料改良9K培养基:(NH4)2SO4 3.5 g, KCl 0.119 g, K2HPO4 0.058 g, Ca(NO3)2·4H2O 0.0168 g, MgSO4·7H2O 0.583 g, 蒸馏水1000 mL, pH 2.5, 121 ℃灭菌30 min.
A. ferrooxidans休止细胞制备:将A. ferrooxidans接种在改良9K培养基中, 置于28 ℃、180 r·min-1摇床中振荡扩培, 指数生长阶段后期停止培养(约2~3 d).将培养液经定性滤纸过滤以除去生成的铁沉淀物, 将滤液以10000×g的相对离心力(4 ℃、10 min)离心收集菌体, 并用pH 1.5的酸水(H2SO4配制)洗3次, 除去各种杂离子.将这些菌体悬浮于pH 2.5的酸水(H2SO4配制), 所得即为A.ferrooxidans浓缩菌液.采用双层平板计数法测得菌液密度为5×108 cell·mL-1.
2.2 试验设置采用去离子水和1:1硫酸配制得到体积为135 mL、pH为2.6、2.3、2.0的若干酸性溶液, 移取15 mL A. ferrooxidans菌液, 使各溶液中A. ferrooxidans密度均为5×107 cell·mL-1.混合均匀后, 准确加入FeSO4·7H2O, 设置Fe2+浓度梯度为20、80和160 mmol·L-1.根据Fe2+浓度调节各体系Na2SO4的加入量, 使得Fe/Na摩尔比分别为0.1、0.5、1.0、2.0, 依此共设置36个处理.将三角瓶置于28 ℃、180 r·min-1摇床中进行生物氧化和水解成矿反应.期间定时观察菌液颜色变化并监测溶液中pH、Fe2+、TFe(总Fe)浓度.待培养至终点时, 观察记录各三角瓶中矿物的表观特征, 通过中速定性滤纸过滤, 并用稀H2SO4 (pH 2.0~3.0)和去离子水分别润洗2次, 以去除矿物表面吸附的离子.收集矿物经35 ℃烘干后进行称重和XRD测定.
2.3 测定方法与数据分析采用pHS-3C精密pH计测定溶液pH值;Fe2+和总Fe浓度采用邻啡罗啉比色法测定;矿物相采用X射线衍射仪测定(XRD, Bruker D8), 测试工作条件为:管电压40 kV, 管电流40 mA, 扫描区间10°~80°(2θ), 步长0.01°, 扫描速率6°·min-1, Cu靶.实验数据采用Origin 8.0软件作图分析.
3 结果与讨论(Results and discussion) 3.1 酸性体系颜色变化和收集矿物的表观特征图 1为160 mmol·L-1 Fe2+体系培养前后颜色的变化情况.因空气氧化Fe2+程度不同导致各酸性体系之间颜色存在差异, 溶液随pH降低由金黄色向浅绿色转变.经生物氧化成矿后, 各体系之间差异愈加明显.在Fe/Na摩尔比同水平下, 溶液最终颜色随初始pH降低而加深, 静置后上清液浊度也随之加大, 且在三角瓶内壁出现结垢现象, 说明合成矿物的颗粒度、沉降性、聚团程度等可能有所不同.从收集矿物的表观特征来看, 矿物颜色随Fe/Na摩尔比的提高而加深, 且pH越低, 形成矿物越少.当pH=2.0、Fe/Na=0.1时, 生物合成的矿物极少, 基本上不能收集检测, 说明在2.0~2.6酸度范围内, 高pH更有利于矿物的生成.
 |
| 图 1 Fe2+浓度为160 mmol·L-1时溶液颜色变化(1~4:pH=2.6, Fe/Na=0.1、0.5、1、2;5~8:pH=2.3, Fe/Na=0.1、0.5、1、2;9~12:pH=2.0, Fe/Na=0.1、0.5、1、2) Fig. 1 Change of solution color under different pH and F/Na molar ratios (Fe2+ initial concentration was 160 mmol·L-1) |
根据图 2可知, Fe2+浓度为160 mmol·L-1时, 各处理间pH的变化过程基本一致, 大致呈先上升后下降的趋势, 并在0~48 h期间出现峰值.这与Fe2+作为能源物质先被A. ferrooxidans氧化(消耗H+, pH升高)和后期Fe3+水解成矿(释放H+, pH降低)密切相关.Fe/Na摩尔比对pH的变化也有一定影响, 表现出Fe/Na摩尔比越大, pH值峰值越小, 培养终点酸度也越大.随着成矿反应的持续进行, Fe/Na摩尔比为0.5、1.0、2.0体系最终pH值分别下降至2.45、2.39、2.32, 而Fe/Na=0.1时则下降幅度较小.分析认为, 出现这种“两极分化”可能有两方面原因:其一, Na2SO4作为中性盐, 当Na+浓度过高时, 由于反应体系渗透压的变化以致菌体水分大量流失而失去活性, A. ferrooxidans氧化Fe2+能力较弱, 间接抑制氧化产物Fe3+的水解成矿过程, 导致pH只升不降或下降幅度较小.其二, 根据反应式(2)、(3)可知, 生物成因施氏矿物在消耗等量Fe3+时所产生的H+比黄铁矾更多, 说明适量Na+的参与可能会改变次生铁矿物的合成行为, 使得Fe3+水解成矿过程更有利于向施氏矿物偏移, 从而导致酸效应过程存在一定的差异.
 |
| 图 2 Fe2+浓度为160 mmol·L-1时pH变化趋势 Fig. 2 Change of solution pH when Fe2+ initial concentration was 160 mmol·L-1 |
图 3为Fe2+浓度在160 mmol·L-1时培养终点收集得到的次生铁矿物XRD图谱.参考JCPDS晶型黄钠铁矾和非晶型施氏矿物的标准图谱可知(Jönsson et al., 2005), 当Na+浓度满足要求时(Fe2+=160 mmol·L-1, Fe/Na=0.5~0.1), 较低的初始pH有利于黄钠铁矾的形成, 表现在pH值由2.6下降至2.0时, 黄钠铁矾的部分衍射峰开始出现并逐渐增强(如2θ=15.84°、17.51°、28.57°、29.10°、40.25°、45.81°、49.68°等).Regenspurg等(2004)把这种酸效应现象归因于体系具备较高的Fe3+供应速率, 从而促进了黄铁矾的形成, 但本试验结果表明并非如此, 在pH=2.0时Fe2+氧化率极低.前已述及, 当Na+浓度在A. ferrooxidans耐受范围内时, 生物成因次生铁矿物的合成过程可能受其浓度高低的影响, 而高浓度Na+则会抑制A. ferrooxidans的Fe2+生物氧化活性, 从而间接阻碍Fe3+的水解成矿过程.因此, 根据反应式(2)、(3)可知, 1 mol Fe3+水解生成施氏矿物可释放出2.75 mol的H+, 而1 mol Fe3+水解生成黄铁矾却只能释放出2 mol的H+.相比之下, 过低的pH值可能对施氏矿物合成行为的抑制作用更大, 从而导致黄钠铁矾更易形成.王长秋等(2005)在用化学方法合成黄钾铁矾时也报道过相似现象.
 |
| 图 3 Fe2+浓度为160 mmol·L-1时收集矿物XRD图谱 Fig. 3 The XRD pattern of minerals when Fe2+ initial concentration was 160 mmol·L-1 |
根据衍射峰位置及强度(2θ=35.16°)判断, 各处理下所获得次生铁矿物中均含有非晶型施氏矿物(Bigham et al., 1994).其中, 当Fe/Na摩尔比为1.0、2.0时, pH在2.0~2.6范围内均收集到纯净施氏矿物.而当Na+浓度上升至320 mmol·L-1时(Fe/Na=0.5), 次生铁矿物的合成途径开始向黄钠铁矾转变, 且其特征衍射峰随着Na+浓度提高愈加明显.Bai等(2012)研究结果表明, 在Fe2+浓度为160 mmol·L-1溶液中, 当K+≥1.60 mmol·L-1(即Fe/K≤100)时就出现黄钾铁矾的衍射峰.而本试验中Fe/Na的摩尔比则需要达到0.1才能实现, 说明Na+的成矾能力远小于K+.
3.4 酸性体系Fe2+、总Fe的变化情况研究考察了初始pH=2.0时, 酸性体系Fe2+浓度和Fe/Na摩尔比对Fe2+氧化和总Fe沉淀去除的综合影响.由图 4可知, 当Na+浓度在A. ferrooxidans耐受范围内时, 160、80、20 mmol·L-1的Fe2+分别在72、48、48 h内被完全生物氧化.当Fe/Na=0.1时, Fe2+浓度为160、80 mmol·L-1处理的生物氧化性均受到抑制, 最终Fe2+氧化率分别只有2.11%和3.61%.而由Fe2+浓度为160 mmol·L-1处理可知, 当Fe/Na=0.5时(Na+浓度320 mmol·L-1), 虽然Fe2+能够在72 h时基本被氧化完全, 但与Fe/Na=2处理相比, 其生物氧化过程相对延迟, 表现在0~24 h内出现微生物延滞期.通过比较可以判断, A. ferrooxidans对Na+的耐受浓度临界值在320~800 mmol·L-1之间, 完全抑制的浓度阈值还有待进一步考察.
 |
| 图 4 初始pH=2.0时Fe2+浓度变化 Fig. 4 Change of Fe2+ concentration in each treatment system when initial pH was 2.0 |
由图 5可知, 当酸性体系pH和Fe2+浓度一定时, 若A. ferrooxidans活性未受到抑制, 则Fe/Na摩尔比在0.5~2.0范围内时对体系总Fe浓度的变化影响较小.这与K+存在较大差异, 柏双友等(2010)发现, 在Fe2+浓度为80 mmol·L-1时, Fe/K摩尔比在25~200之间均能够促使约31%的总Fe以次生铁矿物的形式沉淀去除, 当提高K+浓度至Fe/K=10时, 经生物反应72 h后, 总Fe累积沉淀率超过40%.另外, 160、80、20 mmol·L-1 Fe2+体系的总Fe浓度分别在72、48、48 h时趋于稳定, 至培养终点时平均总Fe沉淀率分别为20.04%、16.43%、0.99%.由此易知, 初始Fe2+浓度越高, Fe3+供应速率越快, 越有利于体系总Fe的沉淀去除.根据Fe3+供应速率v=(x2-x1)/(t2-t1)(式中, x1表示t1时间的Fe3+浓度, x2表示t2时间的Fe3+浓度)可以算出, 在Fe2+被完全氧化时间节点内, 160、80、20 mmol·L-1体系对应的Fe3+供应速率分别为2.22、1.67、0.42 mmol·L-1·h-1, 这也为上述现象提供了合理的解释.进一步分析表明, 总Fe沉淀率的变化趋势与体系Fe2+的氧化过程基本一致, 说明Fe3+的水解过程可能伴随着Fe2+的生物氧化才能进行, 一旦Fe2+氧化完全, 则次生铁矿物的合成也随即终止, 其机理有待进一步研究.
 |
| 图 5 初始pH=2.0时总Fe浓度变化 Fig. 5 Change of total Fe concentration in each treatment system when initial pH was 2.0 |
除了总Fe浓度变化能够表征酸性体系总Fe沉淀去除效果外, 次生铁矿物质量可以更直观地反映A. ferrooxidans生物氧化成矿效果.如图 6所示, 收集矿物质量随着初始pH的降低而出现减少的现象, 这与次生铁矿物的特性有一定的关系.由图 3可知, Na+成矾能力较弱, 各处理产物以施氏矿物居多, 而施氏矿物为典型的球形海胆结构, 易于溶质扩散和矿物溶解(Dold, 2003;Loan et al., 2005;李浙英等, 2011).刘奋武等(2013)在酸性环境下考察了施氏矿物的稳定性, 结果表明, pH体系从3.0下调至2.0后, 施氏矿物在72 h内的溶解率从3.34%猛增至61.46%.依此可以推断, 要使得总Fe沉淀率提高, 对反应体系pH的适当调控至关重要.
 |
| 图 6 培养终点收集矿物质量比较 Fig. 6 The comparison of mineral mass at the end of each treatment |
另外, 从初始Fe2+浓度角度来看, 提高Fe2+浓度有助于矿物的大量合成, 这与图 5结果相吻合.但从Fe/Na摩尔比方面分析, 会发现图 5与图 6之间存在“矛盾”.前已述及, Fe/Na摩尔比在0.5~2.0范围内时对体系总Fe浓度的影响较小, 总Fe沉淀率曲线基本重合.而由图 6则发现在Fe/Na=0.5时能够获取更多的次生铁矿物, 随着Na+浓度减少而呈下降趋势.分析认为, 这与次生铁矿物的合成行为具有一定的关系, 由图 3可知, 当Fe/Na ≥0.5时, 收集矿物开始出现黄钠铁矾衍射峰.而从反应式(2)、(3)来看, 若次生铁矿物按反应式(2)或(3)单独进行时, 则单位Fe3+所合成纯黄铁矾质量应为纯施氏矿物的1.5倍以上, 这也为次生铁矿物产量的增加提供了可能.综上, 虽然Na+成矾能力较弱, 其浓度变化对酸性体系总Fe沉淀去除效果“贡献”较小, 但适量的Na+仍有助于生物成因次生铁矿物的合成.根据反应式(3)可知, Na+的介入可促使酸性硫酸盐环境体系中更多SO42-以次生铁矿物的形式去除.
4 结论(Conclusions)1) Na+浓度在A. ferrooxidans耐受范围内时, 其不影响Fe2+氧化及总Fe沉淀去除效果.160、80、20 mmol·L-1的Fe2+分别在72、48、48 h内被完全氧化, 培养终点时平均总Fe沉淀率分别为20.04%、16.43%、0.99%.
2) 在Fe2+浓度160 mmol·L-1体系中, 当Fe/Na摩尔比为1.0、2.0时, pH在2.0~2.6范围内合成次生铁矿物均为纯净施氏矿物.当Na+浓度提高至320 mmol·L-1时(Fe/Na=0.5), 次生铁矿物的合成途径开始向黄钠铁矾转变, 且其特征衍射峰随着Na+浓度继续提高表现地愈加明显.
柏双友, 梁剑茹, 周立祥. 2011. 一价阳离子和水溶性有机质对生物沥浸中次生铁矿物形成的影响[J]. 矿物学报, 2011, 31(1): 118–125.
|
柏双友, 梁剑茹, 周立祥. 2010. FeSO4-K2SO4-H2O体系中Fe/K摩尔比对生物成因羟基硫酸铁矿物质量的影响及环境意义[J]. 环境科学学报, 2010, 30(8): 1601–1607.
|
Bai S Y, Xu Z H, Wang M, et al. 2012. Both initial concentrations of Fe(Ⅱ) and monovalent cations jointly determine the formation of biogenic iron hydroxysulfate precipitates in acidic sulfate-rich environments[J]. Materials Sciences and Engineering C, 32(8): 2323–2329.
DOI:10.1016/j.msec.2012.07.003
|
Bang S, Patel M, Lippincott L, et al. 2005. Removal of arsenic from groundwater by granular titanium dioxide adsorbent[J]. Chemosphere, 60: 389–397.
DOI:10.1016/j.chemosphere.2004.12.008
|
Bigham J M, Carlson L, Murad E, et al. 1994. Schwertmannite, a new iron oxyhydroxysulfate from Pyhasalmi, Finland, and other localities[J]. Acta Archaeologica, 80(393): 190–192.
|
Bigham J M, Schwertmann U, Traina S J, et al. 1996. Schwertmannite and the chemical modeling of iron in acid sulfate waters[J]. Geochimica et Cosmochimica Acta., 60(12): 2111–2121.
DOI:10.1016/0016-7037(96)00091-9
|
邓志明, 周正华. 2002. 湿法炼锌浸出沉铁探讨[J]. 湖南有色金属, 2002, 18(1): 23–25, 45.
|
Dold B. 2003. Dissolution kinetics of schwertmannite and ferrihydrite in oxidized mine samples and their detection by differential X-ray diffraction (DXRD)[J]. Applied Geochemistry, 18(10): 1531–1540.
DOI:10.1016/S0883-2927(03)00015-5
|
Dutrizac J E. 1996. The effect of seeding on the rate of precipitation of ammonium jarosite and sodium jarosite[J]. Hydrometallurgy, 42: 293–312.
DOI:10.1016/0304-386X(95)00111-S
|
Dutrizac J E, Kaiman S. 1976. Synthesis and properties of jarosite-type compounds[J]. Canadian Mineralogist, 14: 151–158.
|
Dutrizac J E. 1999. The effectiveness of jarosite species for precipitating sodium jarosite[J]. Journal of the Minerals, Metals and Materials Society, 51(12): 30–32.
DOI:10.1007/s11837-999-0168-6
|
Elgersma F, Witkamp G J, Van Rosmalen G M. 1993. Simultaneous dissolution of zinc ferrite and precipitation of ammonium jarosite[J]. Hydrometallurgy, 34: 23–47.
DOI:10.1016/0304-386X(93)90079-S
|
Gan M, Sun S G, Zheng Z H, et al. 2015. Adsorption of Cr(VI) and Cu(Ⅱ) by AlPO4 modified biosynthetic schwertmannite[J]. Applied Surface Science, 356(30): 986–997.
|
Gómez J M, Cantero D, Webb C. 2000. Immobilisation of Thiobacillus ferrooxidans cells on nickel alloy fibre for ferrous sulphate oxidation[J]. Applied Microbiology and Biotechnology, 54: 335–340.
DOI:10.1007/s002530000414
|
Gramp J P, Sandy Jones F, Bigham J M, et al. 2008. Monovalent cation concentrations determine the types of Fe(Ⅲ) hydroxysulfate precipitates formed in bioleach solutions[J]. Hydrometallurgy, 94(1/4): 29–33.
|
Jönsson J, Persson P, Sj berg S, et al. 2005. Schwertmannite precipitated from acid mine drainage:phase transformation, sulfate release and surface properties[J]. Applied Geochemistry, 20: 179–191.
DOI:10.1016/j.apgeochem.2004.04.008
|
Karamanev D G. 1991. Model of the biofilm structure of Thiobacillus ferrooxidans[J]. Journal of Biotechnology, 20(1): 51–64.
DOI:10.1016/0168-1656(91)90034-S
|
Lee W C, Lee S W, Yun S T, et al. 2016. A novel method of utilizing permeable reactive kiddle (PRK) for the remediation of acid mine drainage[J]. Journal of Hazardous Materials, 301: 332–341.
DOI:10.1016/j.jhazmat.2015.09.009
|
Liao Y H, Zhou L X, Liang J R, et al. 2009. Biosynthesis of schwertmannite by Acidithiobacillus ferrooxidans cell suspensions under different pH condition[J]. Materials Science and Engineering:C, 29(1): 211–215.
DOI:10.1016/j.msec.2008.06.011
|
刘奋武, 卜玉山, 田国举, 等. 2013. 温度与pH对生物合成施氏矿物在酸性环境中溶解行为及对Cu2+吸附效果的影响[J]. 环境科学学报, 2013, 33(9): 460–467.
|
Liu G W, Bai R C. 2013. Development of the acidic mining wastewater treatment technology[J]. Applied Mechanics and Materials, 295.
|
李浙英, 梁剑茹, 柏双友, 等. 2011. 生物成因与化学成因施氏矿物的合成、表征及其对As(Ⅲ)的吸附[J]. 环境科学学报, 2011, 31(3): 460–467.
|
Loan M, Richmond W R, Parkinson G M. 2005. On the crystal growth of nanoscale schwertmannite[J]. Journal of Crystal Growth, 275(1/2): 1875–1881.
|
Meschke K, Herdegen V, Aubel T, et al. 2015. Treatment of opencast lignite mining induced acid mine drainage (AMD) using a rotating microfiltration system[J]. Journal of Environmental Chemical Engineering, 4(4): 2848–2856.
|
Mihone K M, Hana F, Sanda R, et al. 2015. Assessment of metal risks from different depths of jarosite tailing waste of Trepça Zinc Industry, Kosovo based on BCR procedure[J]. Journal of Geochemical Exploration, 148: 161–168.
DOI:10.1016/j.gexplo.2014.09.001
|
Regenspurg S, Brand A, Peiffer S. 2004. Formation and stability of schwertmannite in acid mining lakes[J]. Geochimica Et Cosmochimica Acta, 68(6): 1185–1197.
DOI:10.1016/j.gca.2003.07.015
|
Song Y W, Wang M, Liang J R, et al. 2014. High-rate precipitation of iron as jarosite by using a combination process of electrolytic reduction and biological oxidation[J]. Hydrometallurgy, 143(3): 23–27.
|
宋永伟, 赵博文, 霍敏波, 等. 2013. 温度对嗜酸性硫杆菌活性和生物成因次生铁矿物形成的影响[J]. 环境科学, 2013, 34(8): 3264–3271.
|
Vhahangwele M. 2016. A novel technology for neutralizing acidity and attenuating toxic chemical species from acid mine drainage using cryptocrystalline magnesite tailings[J]. Journal of Water Process Engineering, 10(6): 67–77.
|
王长秋, 马生凤, 鲁安怀, 等. 2005. 黄钾铁矾的形成条件研究及环境意义[J]. 岩石矿物学杂志, 2005, 24(6): 607–611.
|
Waychunas G A, Kim C S, Banfield J F. 2005. Nanoparticulate iron oxide minerals in soils and sediments:unique properties and contaminant scanvenging mechanisms[J]. Journa of Nanoparticle Research, 7: 409–433.
DOI:10.1007/s11051-005-6931-x
|
Wei X, Wolfe F A. 2013. Minerals and Mine Drainage[J]. Water Environment Research, 85(10): 1515–1547.
DOI:10.2175/106143013X13698672322507
|
Wu Z L, Zou L C, Chen J H, et al. 2016. Column bioleaching characteristic of copper and iron from Zijinshan sulfide ores by acid mine drainage[J]. International Journal of Mineral Processing, 149: 18–24.
DOI:10.1016/j.minpro.2016.01.015
|
Zhang S L, Jia S Y, Yu B, et al. 2016. Sulfidization of As(V)-containing schwertmannite and its impact on arsenic mobilization[J]. Chemical Geology, 420(20): 270–279.
|
周顺桂, 周立祥, 黄焕忠. 2004. 黄钾铁矾的生物合成与鉴定[J]. 光谱学与光谱分析, 2004, 24(9): 1140–1143.
|
Zhu J Y, Gan M, Zhang D, et al. 2013. The nature of Schwertmannite and Jarosite mediated by two strains of Acidithiobacillus ferrooxidans with different ferrous oxidation ability[J]. Materials Science and Engineering C, 33(5): 2679–2685.
DOI:10.1016/j.msec.2013.02.026
|