 2018, Vol. 38
2018, Vol. 38



NH3选择性催化还原NOx(NH3-SCR)是燃煤电站最为常用的烟气脱硝技术(Busca et al., 1998).而低温SCR技术为当前的研究热点, 这是因为工业锅炉出口的烟气温度往往低于250 ℃, 而燃煤电站常用的V2O5-WO3/TiO2催化剂在该温度区间内活性较低, 无法满足日益严格的NOx排放标准.作为SCR脱硝系统的核心部件, 催化剂的性能对于该系统的脱硝效率起到了决定性作用.国内外研究者发现, Ce基(Peng et al., 2013; Qu et al., 2013; Qu et al., 2016a)、Fe基(Liu et al., 2008; Liu et al., 2010; Liu et al., 2016)、Mn基(Gu et al., 2013; Liu et al., 2013; Jiang et al., 2014)及Cu基(Zhang et al., 2014)催化剂在100~300 ℃温度区间内具有良好的脱硝活性及选择性.然而在低温区间内, 烟气中存在的硫氧化物、水蒸气会不可避免地与还原剂NH3发生反应生成硫酸氢铵堵塞催化剂孔道, 覆盖表面活性位点, 进而严重影响其脱硝性能.因此, 硫酸氢铵的生成为目前制约低温SCR技术工业化应用的主要障碍(Xi et al., 2014).
针对硫酸氢铵这一主要中毒物质, 国内外学者对硫酸氢铵在催化剂表面的形成、分解及与NO反应行为进行了广泛研究.有学者(Zhu et al., 2000; Huang, 2003)发现, 水的加入可加速硫酸氢铵在催化剂表面的沉积;与纯硫酸氢铵相比, 碳基SCR催化剂促进了硫酸氢铵分解及与NO的反应.本课题组之前研究了硫酸氢铵与商业V2O5/TiO2催化剂之间的相互作用, 发现正是由于该相互作用的存在促进了硫酸氢铵在催化剂表面的分解及与NO的反应行为;与结晶态硫酸铵盐相比, 无定形硫酸铵盐更容易分解及与NO反应(Qu et al., 2016b; Ye et al., 2016a; 2016b).因此, 限制硫酸氢铵在催化剂表面过度沉积的一个有效方法就是提高其比表面积来抑制结晶态硫酸铵盐的生成.
基于此, 本文拟考察铁氧化物的添加对硫酸氢铵在催化剂表面分解行为的影响机制.为简化研究, 采用TiO2载体及TiO2-Fe2O3复合载体来研究硫酸氢铵在其表面的分解行为, 同时采用X射线晶体衍射(XRD)、氮吸附、X射线光电子能谱(XPS)、红外光谱及拉曼光谱等实验方法来研究硫酸氢铵与催化剂之间的相互作用.最后通过对V2O5-WO3/TiO2、V2O5-WO3/TiO2-Fe2O3催化剂的活性测试及抗硫抗水性能测试来说明铁氧化物改性的催化剂具有更好的工业应用前景.
2 实验部分(Experimental) 2.1 催化剂制备Fe2O3-TiO2载体的制备:将特定比例的Fe(NO3)3、Ti(SO4)2与0.05 mol·L-1的稀硫酸溶液混合, 60 ℃下水浴搅拌30 min, 即得到澄清溶液;所得溶液在室温下冷却后, 缓慢加入到过量氨水中, 并进行剧烈搅拌;经过滤后, 所得固体用500 mL去离子水洗涤5遍以去除表面残留的SO42-;之后置于烘箱中于110 ℃下干燥过夜, 充分研磨后置于马弗炉中于500 ℃煅烧5 h, 即得到Fe2O3-TiO2(TiFe)载体, 其中, Fe/Ti物质的量比为1:3.
TiO2载体的制备:将Ti(SO4)2与0.05 mol·L-1的稀硫酸溶液混合, 60 ℃下水浴搅拌30 min, 即得到澄清溶液;所得溶液在室温下冷却后, 缓慢加入到过量氨水中, 并进行剧烈搅拌;经过滤后, 所得固体用500 mL去离子水洗涤5遍以去除表面残留的SO42-;之后置于烘箱中于110 ℃下干燥过夜, 充分研磨后置于马弗炉中于500 ℃煅烧5 h, 即得到TiO2(Ti)载体.
大比表面TiO2载体的制备(Chen et al., 2011):将39 g KOH溶于70 mL去离子水, 与2.5 g P25混合后超声20 min, 然后将混合物转移至聚四氟乙烯内胆, 130 ℃水热反应24 h;将所得固体用去离子水洗涤3~5遍以去除表面残留的K+, 并置于1 L 0.1 mol·L-1的稀盐酸溶液中静置6 h;之后将固体用去离子水洗涤3~5遍以去除表面残留的Cl-, 并置于烘箱中于110 ℃干燥过夜, 最后置于马弗炉中于450 ℃煅烧5 h, 即得到大比表面积TiO2载体, 记为Ti-b.
V2O5-WO3/TiO2、V2O5-WO3/TiO2-Fe2O3催化剂的制备(分别简写为VW/Ti及VW/TiFe):将特定量的偏钒酸铵溶于草酸溶液中(偏钒酸铵与草酸物质的量比为1 : 2), 室温下搅拌20 min后静置直到溶液变蓝色, 即得到钒前驱体溶液;将特定量的偏钨酸铵溶于去离子水中, 室温搅拌20 min后即得到钨前驱体溶液.采用过量浸渍法制备催化剂:将上述得到的载体粉末置于500 mL烧瓶中, 并加入特定量的偏钒酸铵溶液与偏钨酸铵溶液, 利用旋转蒸发仪60 ℃水浴加热直到水分完全蒸干;所得固体粉末于烘箱中110 ℃干燥过夜, 并于马弗炉中500 ℃煅烧5 h.经压片、粉碎、研磨及筛分得到40~60目固体颗粒进行活性测试及抗硫抗水性能测试.其中, V2O5及WO3含量分别为3%、5%(质量分数).
硫酸氢铵的负载:将硫酸氢铵溶于去离子水中, 得到硫酸氢铵溶液.将上述得到的载体粉末或者催化剂粉末置于500 mL烧瓶中, 并加入特定量的硫酸氢铵溶液, 利用旋转蒸发仪60 ℃水浴加热直到水分完全蒸干;所得固体粉末于烘箱中110 ℃干燥8 h.经压片、粉碎、研磨及筛分得到40~60目固体颗粒进行活性测试.其中, 硫酸氢铵含量为10%(质量分数), ABS-Ti表示为硫酸氢铵负载于TiO2载体表面.
2.2 催化剂性能评价催化剂活性评价于微反应器中进行, 催化剂用量为0.4 g, 反应气体成分主要为:800 ppm NO、800 ppm NH3、5%O2及N2, 气体流量为1.2 L·min-1, 空速为180000 mL·h-1·g-1.反应气体中的NO及NO2采用在线烟气分析仪(Testo 350)进行检测.NOx(NO+NO2)转化率(η)可用以下公式进行计算:
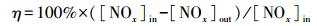
|
(1) |
催化剂抗硫抗水性能于微反应器中进行, 催化剂用量为0.3 g, 反应气体主要为:800 ppm NO、800 ppm NH3、5% O2、250 ppm SO2、5%H2O及N2, 气体流量为1.2 L·min-1, 空速为240000 mL·h-1·g-1, 反应温度为250 ℃.反应气体中的NO及NO2采用在线烟气分析仪(Testo 350)进行检测.NOx转化率用公式(1)进行计算.采用相对活性数值X1/X0来表示催化剂的失活程度, 其中, X0为新鲜催化剂活性, X1为抗硫抗水性能测试过程中某一时刻催化剂活性(Kwon et al., 2015; 2016).
2.3 催化剂表征催化剂XRD表征在D/max-2200 X射线衍射仪(Rigaku公司)上进行, 辐射源为Cu Kα(λ=0.154 nm), 扫描范围2θ=10°~80°, 扫描速率为10°·min-1, 扫描步长为0.02°.采用显微共聚焦拉曼光谱仪(Renishaw, InVia)对样品进行拉曼光谱分析,光谱采集过程中, 分辨率为0.5 cm-1, 累计时间设置为30 s.
XPS在ESCALAB 250 Xi型电子能谱仪上进行, 催化剂表面Ti原子、Fe原子、N原子、S原子的比结合能以C1s(248.58 eV)为标准进行校准.
氮吸附实验在ASAP2020全自动物理吸附仪(Micromeritics公司)中进行, 样品在100 ℃下真空预处理过夜, 在77 K下进行N2吸附, 采用Brunauer-Emmett-Teller(BET)方法计算样品比表面积(SBET), 采用Barrett-Joyner-Halenda(BJH)方法来计算样品孔径分布.
透射红外实验在Nicolet 6700型傅里叶红外光谱仪(Thermo公司)上进行, 首先将样品与KBr混合并充分研磨, 于红外干燥灯下干燥5 min后, 压片即制得检测样品, 扫描次数为32次, 分辨率为4 cm-1.
原位红外实验在Nicolet 6700型傅里叶红外光谱仪(Thermo公司)上进行, 首先将样品于350 ℃下预处理1 h以去除表面吸附的杂质, 同时于100、150 ℃等测试温度点采集背景.反应气体为N2, 气体流量为100 mL·min-1, 扫描次数为64次, 分辨率为4 cm-1.根据所得的原位红外光谱图, 分别计算硫酸根离子及铵根离子的红外峰面积, 同时以100 ℃下硫酸根离子及铵根离子红外特征峰面积为基准, 将150、200、250 ℃下硫酸根离子及铵根离子红外峰面积除以100 ℃下相关官能团红外峰面积, 即得到硫酸根离子及铵根离子的相对含量(Hu et al., 2017).
样品热分析测试在热重分析仪Q500(TA-Q500 TGA)上进行, 称取3 mg固体粉末置于坩埚中, 在氮气气氛下以10 ℃·min-1的升温速率升到100 ℃进行预处理以除去样品表面吸附的水分及其他杂质(处理时间为5 min), 然后以10 ℃·min-1的升温速率升到800 ℃.升温过程中, 天平连续记录样品质量.
3 结果与讨论(Results and discussion) 3.1 硫酸氢铵分解行为研究硫酸氢铵在Ti及TiFe载体上的分解规律如图 1所示.对于ABS-Ti样品而言, 升温过程中出现了3个主要的失重峰(364、455、703 ℃);与高温处的2个失重峰相比较, 位于364 ℃的失重峰较为尖锐, 其DTG值也较高, 说明硫酸氢铵在该温度点具有较高的分解速率.而随着温度的升高, 失重峰所对应的DTG值随之下降, 这可能是由于升温过程中载体表面硫酸铵盐的量下降所导致的.而对于ABS-TiFe而言, 升温过程中出现了4个主要的失重峰(206、394、536、629 ℃), 可以确定Fe的引入改变了硫酸氢铵的分解规律, 促进了其在低温下的分解行为.
 |
| 图 1 TG-DTG曲线(a.ABS-Ti;b.ABS-TiFe) Fig. 1 TG-DTG profiles(a.ABS-Ti; b.ABS-TiFe) |
采用原位红外手段对升温过程中催化剂载体表面铵根离子、硫酸根离子的变化情况进行研究(图 2), 发现随着温度的升高, 铵根离子相对含量明显下降;而且在TiFe载体表面, 铵根离子相对含量的下降幅度明显大于Ti载体, 说明铁氧化物的加入显著促进了表面铵根离子的分解, 这与图 1中的热重结果相吻合.对于硫酸根离子来说, 其相对含量随着温度的变化几乎保持不变, 说明硫酸根离子具有比铵根离子更高的热稳定性.并且对于TiFe载体来说, 位于206 ℃的失重峰可确定为只有铵根离子的分解.而根据升温过程中各样品的质量损失情况及之前的研究可以推断, 350~450 ℃之间的失重峰包含着铵根离子与硫酸根离子的分解, 而高于450 ℃的失重峰则可归结为只有硫酸根离子的分解(Ye et al., 2016a; 2016b).
 |
| 图 2 系列样品中NH4+及SO42-的相对含量 Fig. 2 Relative concentrations of NH4+ and SO42- of the series NH4HSO4-deposited samples |
根据以上结果可以看到, Fe2O3的添加促进了硫酸氢铵的低温分解, 这可能是由于以下原因引起的:①铁氧化物的引入改变了硫酸氢铵中含S、含N官能团在载体表面的存在形式;②硫酸氢铵在TiFe载体表面的附着位点发生变化, 即硫酸根离子除与Ti原子结合之外, 可能还与Fe原子发生相互作用.本文将采用XRD、Raman光谱、氮吸附、红外光谱及XPS手段来探究硫酸氢铵与Ti、TiFe载体之间的相互作用, 进一步揭示硫酸氢铵在系列载体表面的具体存在形态.
3.2 硫酸氢铵与催化剂之间相互作用的研究 3.2.1 物理结构分析采用XRD方法研究样品的晶体结构.如图 3所示, TiO2载体呈现出典型的锐钛矿结构;对于TiFe复合载体而言, 除了出现微弱的锐钛矿TiO2衍射峰之外, 还出现了金红石TiO2衍射峰.这是因为随着铁氧化物的加入, Fe原子会替代部分Ti原子, 从而产生一定的氧空位.考虑到TiO2锐钛矿相与金红石相之间的转变主要与Ti—O键的断裂及Ti、O原子的合作运动有关;而形成的氧空位可为原子排列提供相关空间, 因而促进了TiO2金红石相的生成(Gao et al., 2012).而Fe2O3衍射峰却并未出现, 这可能是由于铁氧化物与钛氧化物形成了固溶体, 或者是由于Fe氧化物的颗粒较小, 分散度较好所致.需要指出的是, 负载硫酸氢铵之后, 并未看到相关硫酸铵盐的衍射峰, 说明硫酸铵盐以无定形的形式存在于催化剂载体表面;并且催化剂载体本身具有的相关衍射峰并未发生相应变化, 说明硫酸氢铵的添加不会对催化剂载体的晶体结构产生相关影响.
 |
| 图 3 系列样品的XRD谱图 Fig. 3 XRD pattern of the series samples |
图 4为系列样品的拉曼光谱图.对于TiO2载体而言, 位于145、197、397、517及639 cm-1处的峰可以归属为锐钛矿TiO2的拉曼振动峰(Jin et al., 2017).对于TiFe复合载体, 除了锐钛矿TiO2的拉曼振动峰之外, 还观测到了归属于Fe2O3(264、304 cm-1)及金红石相TiO2(607 cm-1)的拉曼特征峰(Liu et al., 2010; Frank et al., 2012).同时, 硫酸氢铵加入后, 拉曼峰并没有发生明显变化, 说明硫酸氢铵的引入并未对催化剂载体晶体结构造成明显影响, 这与图 3的XRD结果相吻合.
 |
| 图 4 系列样品的Raman谱图 Fig. 4 Raman spectra of the series samples |
系列样品的比表面积及孔径分布如表 1所示.对于Ti载体来说, 其比表面积较小, 为58 m2·g-1;负载硫酸氢铵后, 其比表面积与孔容有一定程度的降低, 说明硫酸氢铵的沉积堵塞了催化剂载体一部分孔道结构.而添加了Fe2O3之后, 载体比表面积有了巨大的提高, 为208 m2·g-1.结合XRD结果来看, 这可能是因为Fe、Ti原子半径不同, 当Fe原子取代了一部分Ti原子后, TiO2晶格发生了一定程度的畸变, 抑制了晶粒的生长, 降低了晶粒的尺寸, 因而其比表面积有了较为明显的提高(Shan et al., 2012).相似的实验结果在其他学者的研究中可以看到(Gao et al., 2010; Qu et al., 2013).为了消除比表面积对硫酸氢铵分解行为的影响, 制备了大比表面积TiO2载体(其比表面积为250 m2·g-1), 并对其表面硫酸氢铵的分解行为进行了相应探究, 发现比表面积对硫酸氢铵分解行为几乎没有影响(图 5), 而由于Fe2O3的引入导致载体与硫酸氢铵之间相互作用的改变应为硫酸氢铵分解温度降低的主要原因.
| 表 1 系列样品的N2结果 Table 1 N2 adsorption results of the series samples |
 |
| 图 5 ABS-Ti-b的TG-DTG曲线 Fig. 5 TG-DTG profiles of ABS-Ti-b |
采用透射红外光谱来分析硫酸氢铵负载前后催化剂载体表面官能团的存在形式(图 6).对于未负载硫酸氢铵的样品而言, 在1000~2000 cm-1波数段只出现了表面吸附水的红外特征峰(1627 cm-1), 而并未观察到相关硫酸根离子的红外特征峰, 消除了制备过程中载体表面残留的硫酸根离子对之后的研究分析所带来的干扰.Ti载体表面负载硫酸氢铵后, 在1000~2000 cm-1波数段出现了5个红外特征峰, 除了表面吸附水的红外特征峰外, 位于1400 cm-1的峰为铵根离子的振动峰, 而位于1046、1139、1207 cm-1的峰则是处于C2v对称的双齿SO42-的v3振动峰(Xi et al., 2014).其中, 位于1207及1139 cm-1的峰为S O的非对称及对称的伸缩振动峰, 1046 cm-1处的峰则为S=O的非对称伸缩振动峰(Noda et al., 2005; Ropero-Vega et al., 2010).而对于ABS-TiFe样品而言, 依旧可以观察到上述5个红外特征峰.需要值得注意的是, 归属为双齿SO42-的红外特征峰位置出现了一定的红移和蓝移, 说明一部分硫酸根离子很有可能与Fe原子发生了结合, 导致硫酸根离子本身所处的环境发生了某种变化.下面将采用XPS方法来验证该推论.
 |
| 图 6 系列样品的红外光谱图 Fig. 6 FTIR pattern of the series samples |
催化剂载体表面Ti、Fe、N及S原子的XPS曲线如图 7所示.根据Ti 2p1/2及Ti 2p3/2峰的比结合能可以推断Ti原子以+4价的形式存在(图 7a)(Du et al., 2012).而负载硫酸氢铵后, Ti 2p峰向高比结合能方向移动, 说明Ti原子周围的电子云密度有所降低, 硫酸根离子直接与Ti原子连接(Liu et al., 2011).对于TiFe样品, 硫酸氢铵的引入依旧引起了Ti 2p峰比结合能的增加, 说明一部分硫酸根离子依旧与Ti原子结合.
 |
| 图 7 XPS谱图(a.Ti 2p;b.Fe 2p;c.N 1s;d.S 2p) Fig. 7 XPS spectra(a.Ti 2p;b.Fe 2p;c.N 1s;d.S 2p) |
图 7b为Fe 2p XPS曲线图.电子结合能为710.66及724.46 eV处的2个峰分别归属于Fe3+ 2p 3/2及Fe3+2p 1/2, 说明在TiFe复合载体中Fe原子以Fe3+的形式存在(Liu et al., 2010; Liu et al., 2016).负载硫酸氢铵后, Fe 2p峰向高比结合能方向移动, 说明Fe原子处于电子缺失状态, 硫酸根离子除了与Ti原子结合外还与Fe原子进行连接.
图 7c为N 1s XPS谱图.对于ABS-Ti样品来说, 只在401.55 eV处出现了一个XPS光电子峰, 根据之前研究结果, 该峰可归属为NH4+(Ye et al., 2016a).而对于ABS-TiFe样品来说, 经过分峰后除了出现归属于NH4+的XPS峰之外, 还在399.53 eV处出现了一个峰, 经文献查证, 该峰可归属为固体表面吸附态的NH3(Larkins et al., 1979).这可能是因为在XPS测试过程中, 由于真空导致一部分NH4+发生分解, 释放出NH3;而释放的NH3又以氨分子的形式重吸附在固体表面, 从而在399.53 eV处出现了一个新的N 1s XPS峰.这说明NH4+在TiFe表面具有较弱的稳定性, 与图 2中的结果相吻合.
图 7d为S 2p XPS谱图.根据分峰后2个峰的比结合能可知, S原子以硫酸根离子的形式存在(Liu et al., 2011).与ABS-Ti样品相比, TiFe载体上S原子所对应的XPS峰向高比结合能方向发生移动, 说明S原子本身所处的环境发生了相应变化, 这在一定程度上与前文得出的结论相吻合, 即对于TiFe载体, 硫酸根离子除了与Ti原子结合外还与Fe原子连接,而这同样解释了系列载体表面硫酸根离子不同的分解行为.
根据硫酸氢铵热重结果及上述催化剂物理化学表征结果可知, NH4+的稳定性要低于SO42-.而Fe2O3的引入导致一部分硫酸根离子与Fe原子相连, 生成了类似于Fe2(SO4)3的金属硫酸盐, 其热稳定性发生了一定程度的变化.相应的, 硫酸根离子与铵根离子的作用减弱, 因而降低了NH4+消耗所需要的温度.
3.3 催化剂性能研究 3.3.1 催化剂活性研究上文研究了催化剂Ti载体的Fe掺杂改性对硫酸氢铵分解行为的影响规律, 发现TiFe载体的使用可促进硫酸氢铵低温下的分解行为.而活性作为催化剂性能最主要的一个参数, 为决定该催化剂是否具有工业应用潜质的重要指标.本文此处研究了铁氧化物的添加改性对催化剂脱硝活性的影响.如图 8所示, VW/Ti催化剂活性随着温度的提高而提高;当温度达到300 ℃时, 催化剂活性已达到90%以上;进一步提高反应温度, 其活性可接近100%.对于VW/TiFe催化剂而言, 其脱硝活性与VW/Ti催化剂相比并无明显区别.而负载硫酸氢铵后, 催化剂活性有了显著的下降.与负载硫酸氢铵后的VW/Ti催化剂相比, ABS-VW/TiFe样品的活性有了较为明显的提高, 其350 ℃的脱硝活性依旧可以达到95%以上.结合热重结果, 可推测VW/TiFe催化剂可能具有较好的低温抗硫抗水性能.
 |
| 图 8 系列催化剂SCR脱硝活性 Fig. 8 SCR activity of the series catalysts |
催化剂抗硫抗水性能如图 9所示, 其中, 催化剂用量为0.3 g, 反应气体主要为:800 ppm NO、800 ppm NH3、5%O2、250 ppm SO2、5% H2O及N2, 气体流量为1.2 L·min-1, 空速为240000 mL·h-1·g-1, 反应温度为250 ℃.对于VW/Ti催化剂而言, 随着SO2及H2O的加入, 其活性发生了明显的下降;12 h后活性为仅为初始活性的84%左右.对于VW/TiFe催化剂而言, 抗硫抗水性能得到了较为显著的提高, 12 h后活性依旧保留为初始活性的93%左右.这是因为添加的Fe2O3起到了固定硫酸根离子的作用, 促进了硫酸氢铵在低温区间内的分解行为, 一定程度上抑制了硫酸氢铵在催化剂表面的沉积, 从而提高了催化剂的低温抗硫抗水性能.上述结果说明Fe改性的催化剂具有较好的工业应用前景.
 |
| 图 9 催化剂抗硫抗水性能 Fig. 9 Sulfur resistance tests of the series catalysts |
采用共沉淀法合成了TiO2及TiO2-Fe2O3载体, 并研究了硫酸氢铵与上述载体之间的相互作用.结果表明, 在催化剂载体表面, 硫物种以双齿硫酸根离子的形式存在.对于TiO2载体, 硫酸根离子主要与Ti原子相连接;而对于TiO2-Fe2O3复合载体, Ti原子与Fe原子均与硫酸根离子发生相互作用.可知铁氧化物的引入改变了硫酸氢铵与催化剂载体之间的相互作用, 因而促进了硫酸氢铵在低温下的分解行为.催化剂抗硫抗水性能表明, 铁氧化物改性的催化剂具有更好的低温抗硫抗水性能, 为实现低温SCR技术的工业应用提供了良好的理论基础.
Busca G, Lietti L, Ramis G, et al. 1998. Chemical and mechanistic aspects of the selective catalytic reduction of NOx by ammonia over oxide catalysts:A review[J]. Applied Catalysis B:Environmental, 18: 1–36.
DOI:10.1016/S0926-3373(98)00040-X
|
Chen X, Wang H, Wu Z, et al. 2011. Novel H2Ti12O25-confined CeO2 catalyst with remarkable resistance to alkali poisoning based on the "shell protection effect"[J]. The Journal of Physical Chemistry C, 115: 17479–17484.
DOI:10.1021/jp205069w
|
Du X, Gao X, Fu Y, et al. 2012. The co-effect of Sb and Nb on the SCR performance of the V2O5/TiO2 catalyst[J]. Journal of Colloid and Interface Science, 368: 406–412.
DOI:10.1016/j.jcis.2011.11.026
|
Frank O, Zukalova M, Laskova B, et al. 2012. Raman Spectra of titanium dioxide (anatase, rutile) with identified oxygen isotopes (16, 17, 18)[J]. Physical Chemistry Chemical Physics, 14: 14567–14572.
DOI:10.1039/c2cp42763j
|
Gao Q, Wu X, Fan Y. 2012. The effect of iron ions on the anatase-rutile phase transformation of titania (TiO2) in mica-titania pigments[J]. Dyes and Pigments, 95: 96–101.
DOI:10.1016/j.dyepig.2012.03.030
|
Gao X, Du X, Cui L, et al. 2010. A Ce-Cu-Ti oxide catalyst for the selective catalytic reduction of NO with NH3[J]. Catalysis Communications, 12: 255–258.
DOI:10.1016/j.catcom.2010.09.029
|
Gu T, Jin R, Liu Y, et al. 2013. Promoting effect of calcium doping on the performances of MnOx/TiO2 catalysts for NO reduction with NH3 at low temperature[J]. Applied Catalysis B:Environmental, 129: 30–38.
DOI:10.1016/j.apcatb.2012.09.003
|
Hu W, Zhang Y, Liu S, et al. 2017. Improvement in activity and alkali resistance of a novel V-Ce(SO4)2/Ti catalyst for selective catalytic reduction of no with NH3[J]. Applied Catalysis B:Environmental, 206: 449–460.
DOI:10.1016/j.apcatb.2017.01.036
|
Huang Z. 2003. Formation and reaction of ammonium sulfate salts on V2O5/AC catalyst during selective catalytic reduction of nitric oxide by ammonia at low temperatures[J]. Journal of Catalysis, 214: 213–219.
DOI:10.1016/S0021-9517(02)00157-4
|
Jiang B, Deng B, Zhang Z, et al. 2014. Effect of Zr addition on the low-temperature SCR activity and SO2 tolerance of Fe-Mn/Ti catalysts[J]. The Journal of Physical Chemistry C, 118: 14866–14875.
DOI:10.1021/jp412828p
|
Jin Q, Shen Y, Zhu S. 2017. Effect of fluorine additive on CeO2(ZrO2)/TiO2 for selective catalytic reduction of NO by NH3[J]. Journal of Colloid and Interface Science, 487: 401–409.
DOI:10.1016/j.jcis.2016.10.056
|
Kwon D W, Nam K B, Hong S C. 2015. The Role of ceria on the activity and SO2 resistance of catalysts for the selective catalytic reduction of NOx by NH3[J]. Applied Catalysis B:Environmental, 166-167: 37–44.
DOI:10.1016/j.apcatb.2014.11.004
|
Kwon D W, Park K H, Hong S C. 2016. Enhancement of SCR activity and SO2 resistance on VOx/TiO2 catalyst by addition of molybdenum[J]. Chemical Engineering Journal, 284: 315–324.
DOI:10.1016/j.cej.2015.08.152
|
Larkins F P, Lubenfeld A. 1979. The auger spectrum of solid ammonia[J]. Journal of Electron Spectroscopy and Related Phenomena, 15: 137–144.
DOI:10.1016/0368-2048(79)87024-3
|
Liu F, Asakura K, He H, et al. 2011. Influence of sulfation on iron titanate catalyst for the selective catalytic reduction of NOx with NH3[J]. Applied Catalysis B:Environmental, 103: 369–377.
DOI:10.1016/j.apcatb.2011.01.044
|
Liu F, He H, Zhang C. 2008. Novel iron titanate catalyst for the selective catalytic reduction of NO with NH3 in the medium temperature range[J]. Chemical Communications, 17: 2043–2045.
|
Liu F, He H, Zhang C, et al. 2010. Selective catalytic reduction of NO with NH3 over iron titanate catalyst:Catalytic performance and characterization[J]. Applied Catalysis B:Environmental, 96: 408–420.
DOI:10.1016/j.apcatb.2010.02.038
|
Liu F, Shan W, Lian Z, et al. 2013. Novel mnwox catalyst with remarkable performance for low temperature NH3-SCR of NOx[J]. Catalysis Science & Technology, 3: 2699.
|
Liu Z, Su H, Chen B, et al. 2016. Activity enhancement of WO3 modified Fe2O3 catalyst for the selective catalytic reduction of NOx by NH3[J]. Chemical Engineering Journal, 299: 255–262.
DOI:10.1016/j.cej.2016.04.100
|
Noda L K, de Almeida R M, Probst L F D, et al. 2005. Characterization of sulfated TiO2 prepared by the Sol-Gel method and its catalytic activity in the N-Hexane isomerization reaction[J]. Journal of Molecular Catalysis A:Chemical, 225: 39–46.
DOI:10.1016/j.molcata.2004.08.025
|
Peng Y, Liu C, Zhang X, et al. 2013. The effect of SiO2 on a novel CeO2-WO3/TiO2 catalyst for the selective catalytic reduction of NO with NH3[J]. Applied Catalysis B:Environmental, 140-141: 276–282.
DOI:10.1016/j.apcatb.2013.04.030
|
Qu R, Gao X, Cen K, et al. 2013. Relationship between structure and performance of a novel cerium-niobium binary oxide catalyst for selective catalytic reduction of NO with NH3[J]. Applied Catalysis B:Environmental, 142-143: 290–297.
DOI:10.1016/j.apcatb.2013.05.035
|
Qu R, Peng Y, Sun X, et al. 2016a. Identification of the reaction pathway and reactive species for the selective catalytic reduction of NO with NH3 over cerium-niobium oxide catalysts[J]. Catalysis Science & Technology, 6(7): 2136–2142.
|
Qu R, Ye D, Zheng C, et al. 2016b. Exploring the role of V2O5 in the reactivity of NH4HSO4 with NO on V2O5/TiO2 SCR catalysts[J]. RSC Advances, 6: 102436–102443.
DOI:10.1039/C6RA22571C
|
Ropero-Vega J L, Aldana-Pérez A, Gómez R, et al. 2010. Sulfated titania[TiO2/SO42-]:A very active solid acid catalyst for the esterification of free fatty acids with ethanol[J]. Applied Catalysis A:General, 379: 24–29.
DOI:10.1016/j.apcata.2010.02.020
|
Shan W, Liu F, He H, et al. 2012. An environmentally-benign CeO2-TiO2 catalyst for the selective catalytic reduction of NOx with NH3 in simulated diesel exhaust[J]. Catalysis Today, 184: 160–165.
DOI:10.1016/j.cattod.2011.11.013
|
Xi Y, Ottinger N A, Liu Z G. 2014. New insights into sulfur poisoning on a vanadia SCR catalyst under simulated diesel engine operating conditions[J]. Applied Catalysis B:Environmental, 160-161: 1–9.
DOI:10.1016/j.apcatb.2014.04.037
|
Ye D, Qu R, Song H, et al. 2016a. New insights into the various decomposition and reactivity behaviors of NH4HSO4 with NO on V2O5/TiO2 catalyst surfaces[J]. Chemical Engineering Journal, 283: 846–854.
DOI:10.1016/j.cej.2015.08.020
|
Ye D, Qu R, Song H, et al. 2016b. Investigation of the promotion effect of WO3 on the decomposition and reactivity of NH4HSO4 with NO on V2O5-WO3/TiO2 SCR catalysts[J]. RSC Advances, 6: 55584–55592.
DOI:10.1039/C6RA09072A
|
Zhang L, Wang D, Liu Y, et al. 2014. SO2 poisoning impact on the NH3-SCR reaction over a commercial Cu-SAPO-34 SCR catalyst[J]. Applied Catalysis B:Environmental, 156-157: 371–377.
DOI:10.1016/j.apcatb.2014.03.030
|
Zhu Z, Niu H, Liu Z, et al. 2000. Decomposition and reactivity of NH4HSO4 on V2O5/AC catalysts used for NO reduction with ammonia[J]. Journal of Catalysis, 195: 268–278.
DOI:10.1006/jcat.2000.2961
|