 2018, Vol. 38
2018, Vol. 38



硒与人类的血管粥样硬化、克山病、糖尿病、癌症、免疫系统缺陷疾病密切相关(Ambroziak et al., 2017; Ju et al., 2017; Schiavon et al., 2017).湖北恩施、陕西紫阳等地是中国典型的富硒地区, 土壤含量较高的硒可在矿化淋溶、生物等作用下进入水体, 导致水体硒含量过高(刘庆等, 2016).此外, 玻璃、电子、采矿等行业的含硒废水排放也可导致局部水体硒含量升高(Santos et al., 2015).水体中硒含量过高是引起人类硒中毒的主要渠道.吸附方法可有效降低水体中硒的含量, 常用的吸附材料主要有离子交换树脂、活性炭和金属(羟基)氧化物等(Santos et al., 2015).相比树脂和活性炭等材料, 金属(羟基)氧化物等无机矿物表面具有两性羟基(M—OH)和可变电荷, 对硒的吸附容量大且价格低廉、绿色环保而被广泛研究(Santos et al., 2015; Hu et al., 2017; Xia et al., 2017).研究表明, SiO2@氧化铁/氧化铝二元复合物对亚硒酸根(Se(Ⅳ))的吸附容量分别可达20.4 mg·g-1和32.7 mg·g-1(Chan et al., 2009).然而, 金属(羟基)氧化物比表面积不大、孔隙率低, 导致其对硒的吸附性能难以有较大程度的提高(Das et al., 2013; Ma et al., 2018).因此, 有必要研究新材料对水体中硒的吸附特性, 以发展高效吸附水体中硒等污染物的吸附剂.
金属有机骨架材料(MOFs)是一类多孔性、具有不饱和金属位点的聚合物晶体材料.MOFs的巨大比表面积可增大反应物的接触面积, 其不饱和金属原子和官能团可提供大量的活性吸附位点(Martin, 2010; Kumar et al., 2017).因此, MOFs被广泛用于重金属离子、有机染料、生物酶大分子等吸附研究(Wang et al., 2014; Ayati et al., 2016; Lian et al., 2017).尽管MOFs本身具有的不饱和金属位点和官能团均可作为Se(Ⅳ)和Se(Ⅵ)(硒酸根)等含氧阴离子酸根的理想活性吸附位点, 然而国内外将MOFs用于Se(Ⅳ)/Se(Ⅵ)的吸附研究却少见.Howarth等(2015)研究了Uio系列材料吸附Se(Ⅳ)和Se(Ⅵ)的性能, UN-1000对Se(Ⅳ)和Se(Ⅵ)的饱和吸附容量分别为95 mg·g-1和85 mg·g-1, 远高于通常研究的金属(羟基)氧化物吸附剂对二者的饱和吸附容量(Das et al., 2013; Hu et al., 2017; Xia et al., 2017).与Uio系列材料所用的Zr元素相比, Fe元素来源丰富、价格低廉且环境友好.然而, 关于晶体MOF-Fe被用于环境中去除水体Se(Ⅳ)/Se(Ⅵ)的吸附特性研究依然少见, 二者之间的吸附机制研究鲜有报道.此外, 以羧酸基配体合成的MOFs通常在一定pH范围内具有稳定的结构(Burtch et al., 2014), 改变吸附体系的pH可能导致MOFs的结构和性质发生变化, 进而对MOFs的界面物理化学性质和吸附等应用性能及其机制产生影响.Jun等(2015)研究指出, 随着pH从4.1升高至9.2, MIL-100(Fe)对2种有机砷的吸附容量依次降低.Rapti等(2016)研究了Uio-66与海藻酸的复合物(MOR-HA)吸附铬酸根(Cr(Ⅵ))的性能, 发现在pH为1~8的范围内, MOR-HA对Cr(Ⅵ)的去除率随着pH的升高而升高.可见, 吸附体系的pH确实是影响MOFs吸附性能的重要因素.然而, pH如何对MOFs的吸附性能产生影响依旧缺乏明确的研究, 而关于pH影响MOFs吸附Se(Ⅳ)等无机含氧阴离子酸根性能的机制尚无报道.因此, 本研究用水热方法合成了晶体MOF-Fe样品, 对MOF-Fe样品在不同初始pH条件下展开了吸附Se(Ⅳ)的等温吸附和动力学吸附实验, 分析了不同pH条件下MOF-Fe样品吸附Se(Ⅳ)的机制.其结果为深入认识和发展MOFs在环境污染物吸附/分离研究提供基础研究资料.
2 实验部分(Experiment) 2.1 试剂与仪器实验所用试剂均为分析纯级, 超纯水(18.25 MΩ·cm-1)由纯水机(HK-UP-11-20, 浩康科技, 成都)制备.主要仪器有鼓风干燥箱(DHG-9143BS-Ⅲ, 新苗, 上海)、全温培养摇床(QYC-200, 新苗, 上海)、台式高速离心机(CT14DⅡ, 天美, 上海)、X-射线衍射仪(XRD-7000, 岛津, 日本)、氢化物发生-原子荧光分光光度计(AFS-8330, 北京吉天, 中国)、pH计(FE28-Standard, 梅特勒, 瑞士)、激光纳米粒度电位仪(Nano-ZS, 马尔文, 英国)、衰减漫反射红外光谱仪(Agilent cary 600, 安捷伦, 美国)和X-射线光电子能谱仪(ESCALAB 250XI, 赛默飞, 美国).
2.2 试样制备根据文献(Song et al., 2014)的改进方法合成MOF-Fe样品, 主要步骤为:取0.015 mol FeCl3·6H2O(4.05 g)于聚四氟乙烯容器中, 加入75 mL超纯水溶解, 再加入0.01 mol均苯三甲酸(BTC, 2.10 g);将聚四氟乙烯容器放入水热合成反应釜内并置于鼓风干燥箱中, 在150 ℃下合成24 h;自然降至室温, 将产物离心并用超纯水洗涤3次至上清液无色澄清, 再用无水乙醇浸泡4 h并离心2次得到沉淀, 在鼓风干燥箱中80 ℃下烘干得到MOF-Fe样品.
2.3 吸附实验 2.3.1 等温吸附实验用0.01 mol·L-1 KCl溶液配制亚硒酸钠(Se(Ⅳ))溶液为吸附质(2.0 mol·L-1和8.0 mol·L-1).依次将0.01 mol·L-1 KCl溶液和Se(Ⅳ)溶液用1 mol·L-1 NaOH或HCl溶液调节pH至预定值并稳定不变.取若干50 mL塑料离心管, 依次加入不同体积Se(Ⅳ)溶液, 然后补加KCl溶液至30 mL, 再依次准确加入30 mg MOF-Fe粉末, 使吸附体系中Se(Ⅳ)初始浓度依次由0 mol·L-1增大至2.533 mol·L-1, 吸附剂浓度为1 g·L-1.在25 ℃、转速250 r·min-1条件下振荡12 h.振荡结束后, 用pH计测悬浮液的pH, 然后离心得到上清液, 用氢化物发生-原子荧光分光光度计测量上清液中Se(Ⅳ)的浓度.实验重复3次, 取平均值.MOF-Fe样品对Se(Ⅳ)的吸附量(以Se元素计)计算见式(1).
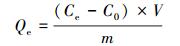
|
(1) |
式中, Qe为MOF-Fe样品对Se(Ⅳ)吸附量(mg·L-1);Ce为Se(Ⅳ)的平衡浓度(mol·L-1);C0为Se(Ⅳ)的初始浓度(mol·L-1);V为吸附体系的体积(L);m为MOF-Fe样品的用量(mg).
2.3.2 动力学吸附实验取90 mL Se(Ⅳ)溶液(1.266 mol·L-1)于100 mL聚四氟乙烯烧杯中, 用油浴锅恒温至25 ℃;在磁力搅拌状态下准确加入90 mg MOF-Fe粉末并开始计时, 分别在时间为0.5、1、2、5、10、20、40、80和160 min时用注射器快速取5 mL悬浮液并用0.22 μm针头过滤器过滤得到上清液, 用氢化物发生-原子荧光分光光度计测量上清液中Se(Ⅳ)的浓度.根据吸附前后溶液中Se(Ⅳ)浓度差值计算样品在不同时间对Se(Ⅳ)的吸附量(以Se元素计).
2.4 吸附机制取等温吸附实验中, 初始pH分别为3.0和5.0、Se(Ⅳ)初始浓度为2.533 mol·L-1实验组吸附平衡的悬浮液, 离心得到沉淀并烘干.对吸附Se(Ⅳ)前后的粉末样品做Zeta电位、衰减漫反射红外光谱(ATR-IR)和X-射线光电子能谱(XPS)测试, 研究MOF-Fe样品吸附Se(Ⅳ)的机制.
根据文献(Petrucci et al., 2002), 亚硒酸的一级、二级电离表达式及平衡常数分别见反应式(2)和式(3):
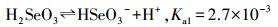
|
(2) |
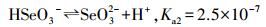
|
(3) |
由反应式(1)和式(2)计算显示, 当pH=3.0时, 体系中[SeO32-]/[HSeO3-]=2.5×10-4, 而[HSeO3-]/[H2SeO3]=2.7;当pH=5.0时, 体系中[SeO32-]/[HSeO3-]=2.5×10-2, 而[HSeO3-]/[H2SeO3]=2.7×102.
2.5 测试方法样品的X-射线衍射(XRD)用X-射线衍射仪测试.测试条件为:Cu靶, 工作电压40 kV、电流30 mA, 步长为0.01°, 扫描速度为8°·min-1.
样品表面Zeta电位用激光纳米粒度电位仪测定.用0.01 mol·L-1 KCl溶液配制0.5 g·L-1样品悬浮液, 超声分散后, 用微量1 mol·L-1 NaOH和HCl溶液在24 h内反复将样品悬浮液的pH分别调至预先设定的值, 取适量悬液测定其Zeta电位.
样品的ATR-IR图谱用衰减漫反射红外光谱仪测试.样品数据采集波数范围550~4000 cm-1, 分辨率2 cm-1.
样品表面XPS测试在X-射线光电子能谱仪上进行.X射线为单色Al Kα(hv =1 486.6 eV)辐射, 功率150 W, 500 μm束斑, 元素扫描步幅0.05 eV, 以C1s(284.6 eV)为标准进行能量校正.用XPSPEAK41软件进行数据处理, 以Shirley方法扣除背景, 采用Lorentzian-Gaussian方法进行分峰拟合.
本研究所有计算、绘图与统计分析均采用Origin8.0软件进行处理.
3 结果与讨论(Results and discussion) 3.1 样品的鉴定MOF-Fe样品的X-射线衍射(XRD)图见图 1.样品的各主要衍射峰的2θ角度分别为3.5°、4.1°、4.9°、6.0°、6.3°、10.3°、11.0°、18.0°、18.7°、19.0°、20.2°和24.1°, 与文献(Song et al., 2014; Guesh et al., 2017)中MIL-100(Fe)的XRD图高度吻合, 表明合成的MOF-Fe样品具有MIL-100(Fe)的晶体结构.根据文献(Kumar et al., 2017), MIL-100(Fe)的结构表达式为FeO3(OH/F)2(H2O)(BTC)2, 其结构单元为(μ3-O2-)连接的3个铁氧八面体.
 |
| 图 1 MOF-Fe样品的X-射线衍射(XRD)图 Fig. 1 XRD of the MOF-Fe sample |
图 2为MOF-Fe样品在不同初始pH条件下对Se(Ⅳ)的等温吸附曲线.3种初始pH条件下, 在Se(Ⅳ)的初始浓度为0~2.533 mol·L-1范围内, 随着Se(Ⅳ)初始浓度的升高样品对Se(Ⅳ)的吸附量(Qe)均表现为先快速增大然后缓慢增加至趋于稳定.当Se(Ⅳ)的平衡浓度相同时, 3种条件下Qe的大小关系均为pH3.0 > pH4.0 > pH5.0.
 |
| 图 2 MOF-Fe样品对Se(Ⅳ)的等温吸附曲线 Fig. 2 Isothermal adsorption curves of Se(Ⅳ) on MOF-Fe sample |
用Langmuir和Freundlich模型对样品吸附Se(Ⅳ)的等温吸附数据进行拟合.2种模型的表达式见式(4)和(5).

|
(4) |
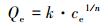
|
(5) |
式中, Qe表示吸附剂对吸附质的平衡吸附量(mol·L-1), ce为吸附体系中吸附质的平衡浓度(mol·L-1).Langmuir模型中Qm表示吸附剂对吸附质的最大吸附容量(mmol·g-1), b为与吸附亲和力及结合能有关的常数(L·mmol-1).Freundlich模型中k为与吸附容量有关的常数(mmol1-(1/n)·L1/n·g-1), 1/n为与吸附强度相关的系数.
表 1为Langmuir和Freundlich模型对样品吸附Se(Ⅳ)的等温吸附数据的拟合参数.Langmuir模型的拟合结果显示, 吸附体系初始pH分别为3.0、4.0和5.0时, MOF-Fe对Se(Ⅳ)的最大吸附容量Qm分别为0.690、0.534和0.538 mmol·g-1;吸附亲和力常数b分别为10.554、5.343和3.138 L·mmol-1, 这说明初始pH为3.0~5.0时, MOF-Fe对Se(Ⅳ)的吸附亲和力随pH升高而降低.根据文献(Hajjaji et al., 2013), Freundlich模型中1/n的大小与吸附强度为负相关;当0.2 < 1/n < 0.5时, 表明吸附剂容易吸附吸附质.3种不同初始pH吸附体系中, Freundlich模型拟合结果1/n均小于0.5且依次增大, 这说明在3种pH体系中Se(Ⅳ)均容易吸附在MOF-Fe上;样品对Se(Ⅳ)的吸附强度大小关系为pH3.0>pH4.0>pH 5.0, 这与Langmuir模型的拟合结果一致.2种模型的拟合度(R2)表明, 在Se(Ⅳ)的初始浓度为0~2.533 mol·L-1范围内, 均质性单分子层吸附的Langmuir模型更适合用于描述样品在3种不同的初始pH吸附体系中MOF-Fe吸附Se(Ⅳ)的特征.
| 表 1 MOF-Fe样品在不同初始pH条件下对Se(Ⅳ)的等温吸附模型拟合参数 Table 1 Fitting parameters of isothermal adsorption models for Se(Ⅳ) adsorbed on MOF-Fe sample at different initial pH* |
图 3为MOF-Fe样品在不同初始pH条件下吸附Se(Ⅳ)的动力学吸附曲线(图 3a)和准二级动力学拟合曲线(图 3b).吸附体系中吸附剂浓度为1 g·L-1, Se(Ⅳ)的初始浓度为1.266 mol·L-1, 吸附体系初始pH分别为3.0、4.0和5.0时, 吸附时间(t)与其对应的吸附量(Qt)与之间的变化关系均表现为:t达到20 min以前, Qt随着t的增加而迅速增大;当t超过20 min时, Qt的增加缓慢并趋于平衡.当t为0.5 min时, 样品在初始pH为3.0、4.0和5.0条件下的Qt分别为0.346、0.318和0.249 mmol·g-1.
 |
| 图 3 MOF-Fe样品对Se(Ⅳ)的动力学吸附曲线(a)和二级动力学方程拟合曲线(b) Fig. 3 Kinetic adsorption curves (a) and fitting curves by pseudo-second order kinetic equation (b) of Se(Ⅳ) on MOF-Fe sample at different initial pH |
用一级动力学速率方程、二级动力学速率方程和Weber-Morris方程(内扩散模型)对MOF-Fe样品吸附Se(Ⅳ)的动力学吸附实验数据进行拟合.一级动力学速率方程、二级动力学速率方程和Weber-Morris方程表达式见式(6)、(7)和(8):
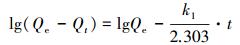
|
(6) |
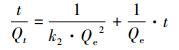
|
(7) |
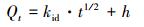
|
(8) |
式中:Qe为样品对吸附质的平衡吸附量(mmol·g-1);Qt为样品在时间t时对吸附质的吸附量(mmol·g-1);k1为一级吸附速率常数(L·min-1);k2为二级吸附速率常数(g·mmol-1·min-1);kid为颗粒内扩散速率常数(mmol·(g·min1/2));h为液膜的厚度.
样品吸附Se(Ⅳ)动力学吸附实验数据的3种模型拟合结果列于表 2.在初始pH分别为3.0、4.0和5.0的吸附体系中, MOF-Fe吸附Se(Ⅳ)的吸附容量与一级动力学速率方程拟合吸附容量不相符, 而与二级动力学速率方程拟合结果相符且R2均大于0.999, 这说明MOF-Fe吸附Se(Ⅳ)的动力学过程可用二级动力学方程描述;且k2分别为2.44、2.17和1.08 g·mmol-1·min-1, 这表明降低吸附体系的pH有助于提高MOF-Fe吸附Se(Ⅳ)的吸附速率.
| 表 2 MOF-Fe样品对Se(Ⅳ)的动力学吸附实验模型拟合参数 Table 2 Fitting parameters of diffusion model and kinetic equation of Se(Ⅳ) by MOF-Fe sample** |
在3种不同初始pH吸附体系中, t1/2与qt之间的显著性水平(p)均小于0.01, 说明样品吸附Se(Ⅳ)的动力学过程与内扩散模型之间存在极显著相关性, 这表明MOF-Fe样品吸附Se(Ⅳ)的过程可用内扩散模型描述.在初始pH分别为3.0、4.0和5.0的吸附体系中, 内扩散模型的拟合参数kid依次增大, 拟合液膜厚度h依次降低.由于MOF-Fe的孔径≤2.5 nm(Song et al., 2014; Guesh et al., 2017), 其吸附活性结构为铁氧八面体结构单元(Jun et al., 2014; Howarth et al., 2015), 属于原子簇层面, 故而不能将MOF-Fe对Se(Ⅳ)的吸附过程看做平板吸附模型;因此MOF-Fe吸附Se(Ⅳ)的液膜厚度可能显著依赖于其铁氧八面体结构中吸附位点处的水合程度相关, 降低吸附体系的pH增大MOF-Fe的水合程度并导致液膜厚度增大.
MOF-Fe样品吸附Se(Ⅳ)的内扩散模型拟合参数表明, 在初始pH为3.0~5.0范围内, 增大吸附体系初始pH可有效提高Se(Ⅳ)在MOF-Fe界面处的内扩散系数;然而二级动力学拟合表明降低吸附体系的pH能有效提高MOF-Fe对Se(Ⅳ)的吸附速率, 这说明改变吸附体系的pH可能导致MOF-Fe吸附Se(Ⅳ)过程中的内扩散速率和化学接触反应吸附速率相对发生了显著地变化.当pH为3.0升高至5.0时, MOF-Fe对Se(Ⅳ)的内扩散速率变快而化学接触反应吸附速率变慢.
4 吸附机制(Mechanism analysis)图 4为MOF-Fe样品吸附Se(Ⅳ)前后的Zeta电位与pH关系曲线.吸附Se(Ⅳ)以前, 样品的等电点(IEP)在pH为3.6附近.当样品悬浮液的pH由3.0升高至5.0时, 样品的Zeta电位由22.0 mV降低至-33.9 mV;悬浮液的pH继续升高至6.0时, Zeta电位反而微略升高.这说明当MOF-Fe悬浮液的pH为5.0~6.0时, 样品中Fe位点被羟基占据完全, 继续增大悬浮液的pH可能导致样品表面的铁羟基不断聚合并形成羟基铁氧化物.吸附Se(Ⅳ)以后, 初始pH为3.0和5.0吸附体系中样品的IEP分别降低至3.4和3.5附近, 说明MOF-Fe吸附Se(Ⅳ)的过程中形成了内圈吸附复合物(Mayordomo et al., 2018).与吸附Se(Ⅳ)以前的样品相比, 吸附Se(Ⅳ)以后样品在悬浮液pH为3.0时的Zeta电位降低, 而在悬浮液pH为5.0时的Zeta电位升高, 这表明静电作用也是MOF-Fe吸附Se(Ⅳ)的机制之一;当pH低于MOF-Fe的IEP时, 样品表面带正电荷与Se(Ⅳ)之间存在的静电引力促进了吸附作用;当pH高于MOF-Fe的IEP时, 样品表面带负电荷与Se(Ⅳ)之间存在的静电排斥作用力不利于二者之间的吸附作用.
 |
| 图 4 样品的pH-Zeta电位图 Fig. 4 Relationship curves of pH-Zeta potential of the samples |
对吸附Se(Ⅳ)前的样品, 以pH分别为3.0、4.0和5.0时的Zeta电位为自变量, 内扩散模型拟合得到的液膜厚度(h)为因变量做相关性分析, 结果显示样品的Zeta电位与样品吸附Se(Ⅳ)过程的h呈显著正相关性(Pearson相关系数为0.9996, p为0.018), 这表明MOF-Fe样品的Zeta电位可以影响MOF-Fe吸附Se(Ⅳ)过程中液膜厚度, 进而影响MOF-Fe吸附Se(Ⅳ)的内扩散过程.综合样品的Zeta电位和样品吸附Se(Ⅳ)内扩散模型拟合结果可知:当吸附体系的pH低于IEP时, 带正电荷的MOF-Fe样品表面发生强烈的水合作用, 形成的扩散层较厚, 导致MOF-Fe吸附Se(Ⅳ)过程中的内扩散系数降低;当吸附体系的pH高于IEP时, 带负电荷的MOF-Fe样品表面水合作用程度减弱, 削弱了铁氧八面体结构中吸附位点表面的扩散层厚度, 导致MOF-Fe吸附Se(Ⅳ)过程中的内扩散系数增大.这与邓东旭等(2017)的研究结果类似.
4.1 初始pH为3.0时图 5为吸附体系初始pH为3.0时MOF-Fe样品吸附Se(Ⅳ)前后的衰减漫反射红外光谱(ATR-IR).根据文献(Cornell et al., 2003; Nakamoto, 2009; Weng, 2010; Silva et al., 2013 Song et al., 2014), 吸附Se(Ⅳ)前MOF-Fe样品ATR-IR图谱中各主要吸收带的归属列于表 3.吸附Se(Ⅳ)以前, 样品分别在1380 cm-1和1449 cm-1处出现了羧基对称伸缩振动吸收带, 在1570 cm-1和1618 cm-1处出现了羧基反对称伸缩振动吸收带;样品中的羧基反对称与对称伸缩振动频率差值分别为238 cm-1和121 cm-1, 说明MOF-Fe中羧基与Fe有单齿和桥式2种键合模式(Weng, 2010; Liu, 2013).根据文献(Cornell et al., 2003; Nakamoto, 2009), 铁氧八面体的铁氧键(Fe—O)变形振动吸收带波数在630 cm-1附近;在pH为3.0时MOF-Fe结构中Fe—O变形振动振动频率增大至640 cm-1, 这说明吸附前在pH为3.0的条件下, MOF-Fe结构中铁羟基(Fe—OH)可能发生质子化作用而脱去羟基形成活性Fe位点.
 |
| 图 5 初始pH为3.0时样品的衰减漫反射红外(ATR-IR)图谱 Fig. 5 ATR-IR spectrums of samples at initial pH 3.0 |
| 表 3 初始pH为3.0时样品的ATR-IR图谱归属列表 Table 3 ATR-IR spectrums absorption band assignments of MOF-Fe*** |
吸附Se(Ⅳ)以后, 样品ATR-IR图谱的变化主要为Fe—O变形振动吸收带波数由640 cm-1降低至625 cm-1, 苯环C—H面外弯曲振动在762 cm-1处的吸收带波数降低至760 cm-1, 在853 cm-1处出现了新的吸收带, 样品结构中单齿键合的羧基C—O吸收带波数由1277 cm-1降低至1273 cm-1, 单齿键合的羧基反对称伸缩振动吸收带波数由1618 cm-1升高至1624 cm-1, 苯环C—H伸缩振动吸收带波数波数由3072 cm-1升高至3084 cm-1, 样品中氢键吸收带强度显著增强且向低波数移动.
图 6为吸附体系初始pH为3.0时MOF-Fe样品吸附Se(Ⅳ)前后表面Fe2p3/2、C1s和Se3d5/2的X-射线光电子能谱(XPS), 吸附Se(Ⅳ)前后Fe2p3/2的分峰拟合数据列于表 4.吸附Se(Ⅳ)以前, 样品中Fe2p3/2的拟合峰可分别归属为:电子结合能(B.E.)值为709.30 eV的拟合峰(Peak1)是由于缺陷位而产生氧化态低的Fe(Grosvenor et al., 2004), B.E.值为711.48 eV的拟合峰(Peak2)对应的是与羧基桥式相连的质子化后的配位饱和Fe, B.E.值为712.96 eV的拟合峰(Peak3)对应的是配位水分子缺失的配位不饱和Fe;B.E.值为716.15 eV的拟合峰(Peak4)可能是样品表面氧化态高的Fe(Grosvenor et al., 2004);B.E.值为718.45 eV的拟合峰(Peak5)是样品中Fe(Ⅲ)2p3/2卫星峰.样品中C1s的拟合峰分别归属为:B.E.值为284.82 eV的拟合峰对应的是苯环sp2 C(Miltenburg et al., 2017), B.E.值为288.83 eV的拟合峰对应的是羧基C(Miltenburg et al., 2017).
 |
| 图 6 初始pH为3.0时样品的X-射线光电子能谱(XPS)图 Fig. 6 XPS spectrums of the samples at initial pH 3.0 |
| 表 4 初始pH为3.0时样品表面Fe2p3/2的XPS分峰数据 Table 4 ata of deconvolution of XPS spectrums for Fe2p3/2 of the samples at initial pH 3.0 |
吸附Se(Ⅳ)以后, 样品Fe2p3/2图谱中Peak2含量增多, 而Peak3含量减少, 二者的比值由0.522升高至1.06;Peak4消失, Peak5峰位B.E.值降低至717.09 eV, 半峰宽(FWHM)增大了4.09 eV, 这可能是Peak4和Peak5不能有效分离所致.羧基C1s的B.E.值增大了0.03 eV.吸附态Se3d5/2出现了2个拟合峰, 其B.E.值分别为59.01 eV和59.98 eV.
当吸附体系的pH为3.0时, Se(Ⅳ)主要以[O2SeOH]-和和H2SeO3形态存在.根据样品吸附Se(Ⅳ)前后的ATR-IR和XPS图谱可知, ①吸附Se(Ⅳ)以后, MOF-Fe结构中Fe—O变形振动频率降低, ATR-IR图谱中在853 cm-1处出现了Fe-O-Se振动吸收带(Nakamoto, 2009), 且样品Fe2p3/2 XPS图谱中Peak2的B.E.值略有降低, 而吸附态Se3d5/2在59.01 eV出现了拟合峰(注:对亚硒酸钠和硒酸钠做了XPS测试, 其Se3d5/2的B.E.值分别为58.68 eV和60.14 eV);这表明初始pH为3.0的吸附体系中羟基质子化后暴露的活性Fe原子与[O2SeOH]-形成了Fe—O—Se结构.②吸附Se(Ⅳ)以后的样品Fe2p3/2 XPS图谱中Peak3含量减少而Peak2含量增大, 吸附态Se3d5/2在B.E.值为59.98 eV处出现了拟合峰;这表明H2SeO3形态的Se原子与配位不饱和Fe原子发生配位作用, 导致配位不饱和Fe原子电子云密度增大而Se原子电子云密度降低(Liu, 2013).③吸附Se(Ⅳ)以后, 样品结构中单齿键合的羧基反对称伸缩振动吸收带波数增大, 羧基C—O吸收带波数降低;这可能是单齿键合的羧羰基氧与Se(Ⅳ)中氢原子之间形成氢键作用所致.Se(Ⅳ)与羰基氧的氢键作用促使羧基C电子云向氧原子偏移, 导致羧基C1s的B.E.值增大;羧基官能团电子云的偏移或吸附态Se(Ⅳ)对苯环大п键产生了弱的诱导效应, 致使样品的苯环C—H面外弯曲振动吸收带波数略有降低, 而C—H伸缩振动吸收带波数增大.此外, Se(Ⅳ)与羧基之间形成的氢键作用也可导致吸附Se(Ⅳ)以后样品的氢键振动吸收带增强;H2SeO3与样品形成配位吸附态Se(Ⅳ), 促进了体系中Se(Ⅳ)的一级电离平衡向左进行, 导致体系pH升高.
4.2 初始pH为5.0时图 7为吸附体系初始pH为5.0时MOF-Fe样品吸附Se(Ⅳ)前后的ATR-IR图谱.吸附Se(Ⅳ)以前, 与初始pH为3.0样品的ATR-IR图谱相比, 吸附体系初始pH为5.0时样品Fe—O变形振动吸收带波数由640 cm-1降低至629 cm-1, 样品中单齿键合的羧基反对称吸收峰波数由1618 cm-1升高至1620 cm-1, 苯环C—H伸缩振动吸收带波数升高至3087 cm-1, 在3224 cm-1处出现了配位水分子的振动吸收带(Silva et al., 2013), 且在3366 cm-1处出现了氢键强烈吸收带.
 |
| 图 7 初始pH为5.0时样品的ATR-IR图谱 Fig. 7 ATR-IR spectrums of the samples at initial pH 5.0 |
吸附Se(Ⅳ)以后, 样品ATR-IR图谱的主要变化为Fe—O变形振动吸收带继续降低至626 cm-1, 苯环C—H在波数为761 cm-1处面外弯曲振动吸收带波数降低至759 cm-1, 样品在850 cm-1附近出现了吸附态Fe—O—Se吸收带, 在1045 cm-1处出现了C—O振动吸收带, 单齿键合的羧基C—O吸收带波数由1279 cm-1降低至1271 cm-1, 单齿键合的羧基反对称伸缩振动吸收带波数继续升高至1624 cm-1, 3366 cm-1处的氢键吸收带强度增强.
图 8为吸附体系的初始pH为5.0时MOF-Fe样品吸附Se(Ⅳ)前后表面Fe2p3/2、C1s和Se3d5/2的XPS, 吸附Se(Ⅳ)前后Fe2p3/2的分峰拟合数据列于表 5.MOF-Fe样品吸附Se(Ⅳ)以后, 配位饱和Fe原子和不饱和Fe原子的相对含量变化不大, 两种Fe2p3/2的B.E.值分别降低了0.07 eV和0.10 eV;Fe(Ⅲ)2p3/2出现了2个卫星峰, 相应的B.E.值分别为717.30 eV和719.83 eV.苯环sp2 C1s的B.E.值降低了0.05 eV, 而羧基C1s的B.E.值增大了0.03 eV, 且羧基C1s拟合峰的FWHM增大了0.22 eV.样品中吸附态Se(Ⅳ)3d5/2出现了2个拟合峰, 其B.E.值分别为58.84 eV和59.76 eV.
 |
| 图 8 初始pH为5.0时样品的XPS图 Fig. 8 XPS spectrums of the samples at initial pH 5.0 |
| 表 5 初始pH为5.0时样品表面Fe2p3/2的XPS分峰数据 Table 5 Data of deconvolution of XPS spectrums for Fe2p3/2 of the samples at initial pH 5.0 |
当吸附体系初始pH为5.0时, 尽管Se(Ⅳ)主要以[O2SeOH]-形式存在, 但[SeO3]2-形态的含量增大.样品吸附Se(Ⅳ)以后, ① Fe—O变形振动吸收带波数继续降低至626 cm-1, 并在850 cm-1附近出现了Fe—O—Se振动吸收带, 这说明吸附体系中Se(Ⅳ)与样品中的Fe—OH与发生阴离子交换作用;Se(Ⅳ)与Fe—OH之间的阴离子交换作用导致吸附态Se3d5/2的XPS图谱中出现了B.E.值为58.84 eV的拟合峰, 而羟基交换位点Fe2p3/2的B.E.值减小.②样品ATR-IR图谱中出现了非共轭态的C—O振动吸收带, 说明吸附体系中Se(Ⅳ)破坏了桥式羧羟基Fe—O键.尽管吸附态Se3d5/2出现了B.E.值为59.76 eV的拟合峰, 但吸附Se(Ⅳ)以后样品中饱和/不饱和Fe的相对含量并没有明显的变化, 且样品中配位水分子的振动吸收带没有减弱, 结合样品吸附Se(Ⅳ)前后的ATR-IR和XPS图谱的变化可推断:在初始pH为5.0的吸附体系中, [SeO3]2-破坏桥式羧羟基Fe—O键的同时并取代了Fe—OH, 与Fe形成了双齿键合吸附态Se(Ⅳ);吸附体系中[SeO3]2-含量减少, 促进了体系中Se(Ⅳ)的电离平衡向右进行, 导致体系的pH降低.此外, 样品中单齿键合的羧基反对称伸缩振动吸收带波数增大, 羧基C—O吸收带波数降低;这表明在初始pH为5.0的吸附体系中, 单齿键合的羧羰基氧与Se(Ⅳ)中氢原子之间氢键作用仍是二者的吸附机制之一.
通过研究初始pH分别为3.0和5.0的吸附体系中MOF-Fe吸附Se(Ⅳ)前后样品的ATR-IR和XPS可知, MOF-Fe样品对Se(Ⅳ)的吸附机制如图 9所示, 当吸附体系初始pH为3.0时, MOF-Fe对Se(Ⅳ)的化学吸附机制主要为:①质子化脱羟基形成的活性Fe与[O2SeOH]-之间的键合作用, ②配位水分子脱去形成的配位不饱和Fe与H2SeO3之间的配位作用, ③ Se(Ⅳ)氢原子与单齿键合的羧羰基氧之间的氢键作用.当吸附体系初始pH为5.0时, MOF-Fe中的Fe—OH与[O2SeOH]-发生阴离子交换作用;[SeO3]2-破坏桥式羧羟基Fe—O键后与Fe形成了双齿键合吸附态Se(Ⅳ);此外, 也存在Se(Ⅳ)氢原子与单齿键合的羧羰基氧之间的氢键作用.
 |
| 图 9 MOF-Fe吸附Se(Ⅳ)的机制 Fig. 9 Mechanisms of Se(Ⅳ) adsorbed on MOF-Fe |
对MOF-Fe吸附Se(Ⅳ)的机制分析表明, 吸附体系的初始pH影响MOF-Fe吸附Se(Ⅳ)的主要原因为:①低pH导致MOF-Fe表面的Fe—OH和配位水分子发生质子化作用而成为Fe—OH的Brønsted共轭酸位点和Lewis酸位点等吸附位点, 而高pH则导致MOF-Fe表面只存在Fe—OH等Brønsted碱活性吸附位点, 且质子化作用后形成的Brønsted共轭酸位点和Lewis酸位点有利于强化表面水合作用(邓东旭等, 2017), 这与Zeta电位分析结论一致;②改变吸附体系的pH, 导致Se(Ⅳ)的赋存形态发生变化, 进而改变MOF-Fe吸附Se(Ⅳ)的机制.
5 结论(Conclusions)1) 初始pH分别为3.0、4.0和5.0时, Langmuir模型可较好的描述MOF-Fe样品对Se(Ⅳ)的等温吸附数据;降低吸附体系的初始pH可有效提高MOF-Fe样品对Se(Ⅳ)的吸附亲和力和最大吸附容量.3种初始pH不同的体系中, MOF-Fe对Se(Ⅳ)的吸附过程均符合二级动力学方程;但在吸附Se(Ⅳ)的过程中, 其内扩散速率和化学接触反应速率受吸附体系的pH影响明显, 增大pH导致MOF-Fe对Se(Ⅳ)的内扩散速率变快而化学接触反应吸附速率变慢.
2) 当吸附体系初始pH为3.0时, MOF-Fe发生质子化作用而形成Lewis酸和Brønsted共轭酸等活性吸附位点, 与Se(Ⅳ)的化学吸附主要包括Lewis酸位点与H2SeO3之间的配位和Brønsted共轭酸位点与[O2SeOH]-之间的键合等吸附作用机制.当吸附体系初始pH为5.0时, MOF-Fe中只有Fe—OH等Brønsted碱活性吸附位点, Fe—OH与[O2SeOH]-发生阴离子交换作用和[SeO3]2-破坏桥式羧羟基Fe—O键后与Fe形成了双齿键合吸附态Se(Ⅳ)是其主要吸附机制.可见, 吸附体系的初始pH通过影响MOF-Fe的活性吸附位点形式、密度以及Se(Ⅳ)的形态进而影响MOF-Fe样品对Se(Ⅳ)的吸附性能.
Ayati A, Shahrak M N, Tanhaei B, et al. 2016. Emerging adsorptive removal of azo dye by metal-organic frameworks[J]. Chemosphere, 160: 30–44.
DOI:10.1016/j.chemosphere.2016.06.065
|
Ambroziak U, Hybsier S, Shahnazaryan U, et al. 2017. Severe selenium deficits in pregnant women irrespective of autoimmune thyroid disease in an area with marginal selenium intake[J]. Journal of Trace elements in Medicine and Biology, 44: 186–191.
DOI:10.1016/j.jtemb.2017.08.005
|
Burtch N C, Jasuja H, Walton K S. 2014. Water Stability and Adsorption in Metal-Organic Frameworks[J]. Chemical Reviews, 114(20): 10575–10612.
DOI:10.1021/cr5002589
|
Cornell R M, Schwertmann U. 2003. The iron oxides, structure, properties, reactions occurences, uses, 2nd ed[M]. Weinheim, Germany: Wiley-VCH.
|
Chan Y T, Kuan W H, Chen T Y, et al. 2009. Adsorption mechanism of selenate and selenite on the binary oxide systems[J]. Water Research, 43(17): 4412–4420.
DOI:10.1016/j.watres.2009.06.056
|
邓东旭, 王磊, 李兴飞, 等. 2017. 超滤过程中蛋白质带电性对水合作用的影响机制[J]. 哈尔滨工业大学学报, 2017, 49(8): 78–82.
DOI:10.11918/j.issn.0367-6234.201608035 |
Das S, Jim H M, Essilfie-Dughan J. 2013. Adsorption of selenate onto ferrihydrite, goethite, and lepidocrocite under neutral pH conditions[J]. Applied Geochemistry, 28: 185–193.
DOI:10.1016/j.apgeochem.2012.10.026
|
Grosvenor A P, Kobe B A, Biesinger M C, et al. 2004. Investigation of multiplet splitting of Fe2p XPS spectra and bonding in iron compounds[J]. Surf Interface Anal, 36(12): 1564–1574.
DOI:10.1002/(ISSN)1096-9918
|
Guesh K, Caiuby C A D, Mayoral Á, et al. 2017. Sustainable preparation of MIL-100(Fe) and its photocatalytic behavior in the degradation of methyl orange in water[J]. Crystal Growth & Design, 17(4): 1806–1813.
|
Howarth A J, Katz M J, Wang T C, et al. 2015. High Efficiency Adsorption and Removal of Selenate and Selenite from Water using Metal-Organic Frameworks[J]. Journal of the American Chemical Society, 137: 7488–7484.
DOI:10.1021/jacs.5b03904
|
Hu B W, Ye F, Jin C G, et al. 2017. The enhancement roles of layered double hydroxide on the reductive immobilization of selenate by nanoscale zero valent iron:Macroscopic and microscopic approaches[J]. Chemosphere, 184: 408–416.
DOI:10.1016/j.chemosphere.2017.05.179
|
Hajjaji W, Ganiyu S O, Tobaldi D M, et al. 2013. Natural portuguese clayey materials and derived TiO2-containing composites used for decolouring methylene blue (MB) and orange Ⅱ (OⅡ) solutions[J]. Applied Clay Science, 83-84: 91–98.
DOI:10.1016/j.clay.2013.08.013
|
Ju W, Li X, Li Z, et al. 2017. The effect of selenium supplementation on coronary heart disease:A systematic review and meta-analysis of randomized controlled trials[J]. Journal of Trace elements in Medicine and Biology, 44: 8–16.
DOI:10.1016/j.jtemb.2017.04.009
|
Jun J W, Tong M M, Jung B K, et al. 2015. Effect of central metal ions of analogous metal-organic frameworks on adsorption of organoarsenic compounds from water:Plausible mechanism of adsorption and water Purification[J]. Chemistry-A European Journal, 21: 347–354.
DOI:10.1002/chem.201404658
|
Kumar P, Pournara A, Kim K H, et al. 2017. Metal-organic frameworks:Challenges and opportunities for ion-exchange/sorption applications[J]. Progress in Materials Science, 86: 25–74.
DOI:10.1016/j.pmatsci.2017.01.002
|
刘庆, 田侠, 史衍玺. 2016. 外源硒矿粉对玉米硒累积及矿质元素吸收的影响植物[J]. 营养与肥料学报, 2016, 22(2): 403–409.
DOI:10.11674/zwyf.14351 |
刘伟生. 2013. 配位化学[M]. 北京: 化学工业出版社.
|
Lian X Z, Fang Y, Joseph E, et al. 2017. Enzyme-MOF (metal-organic framework) composites[J]. Chemical Society Reviews, 46: 3386–3401.
DOI:10.1039/C7CS00058H
|
Miltenburg M B, Schon T B, Kynaston E L, et al. 2017. Electrochemical polymerization of functionalized graphene quantum Dots[J]. Chemistry of Materials, 29(16): 6611–6615.
DOI:10.1021/acs.chemmater.7b01700
|
Mayordomo N, Foerstendorf H, Lützenkirchen J, et al. 2018. Selenium(Ⅳ) sorption onto γ-Al2O3:A consistent description of the surface speciation by spectroscopy and thermodynamic modeling[J]. Environmental Science & Technology, 52(2): 581–588.
|
Martin S. 2010. Functional Metal-Organic Frameworks: Gas Storage, Separation and Catalysis[M]. Springer
|
Ma Z Y, Shan C, Liang J L, et al. 2018. Efficient adsorption of Selenium(Ⅳ) from water by hematite modified magnetic nanoparticles[J]. Chemosphere, 193: 134–141.
DOI:10.1016/j.chemosphere.2017.11.005
|
Nakamoto K. 2009. Infrared and Raman spectra of inorganic and coordination compounds, 6nd ed[M]. John Wiley & Sons, Inc
|
Petrucci R H, Harwood W S, Herring F G. 2002. General chemistry:Principles and modern applications, 8nd ed[M]. Upper Saddle River, NJ: Prentice Hall.
|
Rapti S, Pournara A, Sarma D, et al. 2016. Rapid, green and inexpensive synthesis of high quality UiO-66 amino-functionalized materials with exceptional capability for removal of hexavalent chromium from industrial waste[J]. Inorganic Chemistry Frontiers, 3: 625–644.
|
Song G Q, Wang Z Q, Wang L, et al. 2014. Preparation of MOF(Fe) and its catalytic activity for oxygen reduction reaction in an alkaline electrolyte[J]. Chinese Journal of Catalysis, 35(2): 185–195.
DOI:10.1016/S1872-2067(12)60729-3
|
Silva I G N, Kai J, Felinto M C F C, et al. 2013. White emission phosphors based on Dy3+-doped into anhydrous rare-earth benzenetricarboxylate complexes[J]. Optical Materials, 35(5): 978–982.
DOI:10.1016/j.optmat.2012.12.001
|
Schiavon M, Ertani A, Parrasia S, et al. 2017. Selenium accumulation and metabolism in algae[J]. Aquatic Toxicology, 189: 1–8.
DOI:10.1016/j.aquatox.2017.05.011
|
Santos S, Ungureanu G, Boaventura R, et al. 2015. Selenium contaminated waters:An overview of analytical methods, treatment options and recent advances in sorption methods[J]. Science of the Total Environment, 521-522: 246–260.
DOI:10.1016/j.scitotenv.2015.03.107
|
翁诗甫. 2010. 傅里叶变换红外光谱分析[M]. 北京: 化学工业出版社.
|
Wang Y, Xie J, Wu Y C, et al. 2014. A magnetic metal-organic framework as a new sorbent for solid-phase extraction of copper(Ⅱ), and its determination by electrothermal AAS[J]. Microchim Acta, 181(9/10): 949–956.
|
Xia X, Ling L, Zhang W. 2017. Solution and surface chemistry of the Se(Ⅳ) -Fe(0) reactions:Effect of initial solution pH[J]. Chemosphere, 168: 1597–1603.
DOI:10.1016/j.chemosphere.2016.11.150
|