 2018, Vol. 38
2018, Vol. 38



化石燃料在推动工业社会向前发展的同时, 其燃烧过程也排放了大量的污染物, 导致严重的环境和健康问题, 尤其是氮氧化物(主要是一氧化氮(NO)和二氧化氮(NO2))与水分结合产生的HNO3(酸雨), 增加了呼吸系统疾病的风险.因此, 控制氮氧化物排放迫在眉睫.目前, 主要的NOx控制方法包括NOx的催化还原法和催化氧化法, 这些方法都被视为控制NOx排放的有效方法(Mochida et al., 1994;Kyotani et al., 1999).
不论是催化氧化还是催化还原NOx, 催化剂都是其核心.氧化石墨烯(Graphene oxide, GO)因具有独特的结构和性能, 而常被用作制备脱硝催化剂的重要载体.GO通常由石墨与氧化剂在溶液中反应合成(Liu et al., 2014;Han et al., 2014), 因此, GO含有沿着二维碳晶格无序分布的环氧化物和羟基官能团.这些基团与平面之间以共价键相连, 与晶格中的碳原子以sp3杂化键成键(Dreyer et al., 2009;Hummers et al., 1958;Szabo et al., 2006;Hernandez et al., 2008;Schniepp et al., 2006;Acik et al., 2010;Robinson et al., 2008;Park et al., 2008;Robinson et al., 2008;Lerf et al., 1998).GO一般可通过强氧化方法制得, 例如, Hummers方法常导致石墨片层碳空位缺陷, 易形成羟基化石墨烯(hydroxylated graphene, HyG)和羧基化石墨烯(carboxylated graphene, CyG) (Hummers et al., 1958).其中, —OH基团可以促进电子转移和NO吸附, 例如, Bi/GO纳米杂化物在280 nm光照下具有氧化NO的光催化活性, 增强了光生载流子的分离和转运, TiO2/ZnO/Bi2O3-GO体系在太阳光照射下对NO降解表现出较好的光催化活性.由此可见, 利用羟基来修饰石墨烯具有重要的意义.
HyG基对催化剂催化降解NO具有协同增效作用(Wang et al., 2017).但有关烟气脱硝过程中HyG对NO的氧化反应特性尚缺乏研究.因此, 本文基于密度泛函理论(Density Functional Theory, DFT)计算, 研究HyG直接氧化NO的反应活性.同时, 根据不同石墨烯超晶胞5×5×1、6×6×1、7×7×1、8×8×1和9×9×1的优化结果对HyG进行建模, 研究HyG复合材料的电子特性和氧化还原性能, 并进行热力学和动力学分析, 讨论HyG氧化NO的反应特性.以期为氧化石墨烯基催化剂的优化设计提供指导.
2 建模和计算细节(Modelling and computational details)以单层石墨碳原子层为基体, 构建5×5×1、6×6×1、7×7×1、8×8×1和9×9×1等周期性超晶胞表面模型, 即G(5×5)、G(6×6)、G(7×7)、G(8×8)和G(9×9).然后在G(5×5)、G(6×6)、G(7×7)、G(8×8)和G(9×9)基础上, 分别去掉一个碳原子, 优化获得碳原子缺陷超表面, 即:Gdef.(5×5)、Gdef.(6×6)、Gdef.(7×7)、Gdef.(8×8)和Gdef.(9×9).基于理想和碳缺陷的石墨烯超表面, 用—OH与表面键合进行几何优化, 分别得到羟基化的石墨烯HyG超表面模型:HO-G(5×5)、HO-G(6×6)、HO-G(7×7)、HO-G(8×8)、HO-G(9×9)、HO-Gdef(5×5)、HO-Gdef(6×6)、HO-Gdef(7×7)、HO-Gdef(8×8)和HO-Gdef(9×9), 进行几何优化获得稳定的模型, 具体如图 1所示.为了得到HyG复合材料与NO反应的反应机理, 进一步研究了稳定HyG模型表面上NO分子的吸附和氧化反应, 所有计算均使用DMol3模块进行.由于LDA(Local Density Approximation)近似具有较强的电子分布局部化, 不适合分析禁带宽度和结合能的绝对值, 因此, DFT计算由具有PBE参数化的交换相关电位的GGA (Generalized Gradient Approximation)完成(Payne et al., 1992).采用双数值加d函数基组, 用超软赝势描述离子核(Gao et al., 2017).为了减少NO或NO2分子之间的相互作用, 用周期性条件模拟石墨烯和GO上NO的吸附.参考以前关于界面相互作用的工作(Li et al., 2016;Qin et al., 2015), 为了避免相邻石墨烯原子层之间的干扰, 将超晶胞向上和向下延伸1.5 nm.在几何优化中, 所有原子都不被固定, 直到任何原子上的所有力分量都小于1×10-5 Ha·nm-1.自洽场中能量、最大力、最大应力和位移的收敛标准值分别为2.0×10-5 eV·atom-1、0.0002 Ha·nm-1、0.1 GPa和0.0002 nm.使用线性同步传输或二次同步传输方法(Linear Synchronous Transit/Quadratic Synchronous Transit, LST/QST)(Halgren et al., 1977; Liang et al., 2017.)来搜索被NO还原的HyG的过渡状态(TS).
 |
| 图 1 HO-G(5×5)、HO-G(6×6)、HO-G(7×7)、HO-G(8×8)、HO-G(9×9)、HO-Gdef.(5×5)、HO-Gdef.(6×6)、HO-Gdef.(7×7)、HO-Gdef.(8×8)、和HO-Gdef.(9×9)的稳定结构 Fig. 1 Stable configurations of the HO-G(5×5), HO-G(6×6), HO-G(7×7), HO-G(8×8), HO-G(9×9), HO-Gdef.(5×5), HO-Gdef (6×6), HO-Gdef (7×7), HO-Gdef(8×8), and HO-Gdef(9×9) hydroxylated composites |
为了避免HyG模型(表示为HO-G)的边界效应(Heyrovska et al., 2008;Castro et al., 2010;Wallace et al., 1947), 需构建大小适合的超晶胞.石墨烯超晶胞5×5×1、6×6×1、7×7×1、8×8×1和9×9×1的C—C键长度为0.1420~0.1421 nm, 与实验值一致.
—OH基团结合石墨烯的结合能(Ebind)按照下式计算:

|
(1) |
式中, E(HO-G)是HO-G模型的总能量(eV);E(G)和E(OH)分别表示石墨烯的总能量和—OH基团的能量(eV).根据式(1)计算得出—OH基团结合到5×5×1、6×6×1、7×7×1、8×8×1和9×9×1石墨烯超晶胞的Ebind分别为-0.708、-1.001、-0.727, -0.781和-0.914 eV(表 1).可见, 6×6×1石墨烯超晶胞中的—OH基团和石墨烯之间的结合是最稳定的.
| 表 1 计算的Edef、Ebind(HO-G)和Ebind (HO-Gdef) Table 1 The calculated Edef, Ebind(HO-G), andEbind(HO-Gdef) |
作为比较, 5×5×1、6×6×1、7×7×1、8×8×1和9×9×1的缺陷型超晶胞的碳空位缺陷形成能Edef.定义如下:
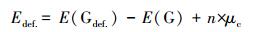
|
(2) |
式中, E(Gdef)和E(G)分别是缺陷和无缺陷的石墨烯超晶胞系统的总能量(eV); μc是碳原子的化学势即石墨烯中每个原子的能量; n是去除原子空位(正值)或添加吸附原子(负值)的数量.
碳原子空位使石墨烯原子层发生Jahn-Teller畸变, 破坏石墨烯原子层的对称性(El-Barbary et al., 2003;Telling et al., 2003), 其中, 靠近碳原子空位的3个C原子之间的距离分别为0.257、0.257和0.202 nm.从表 1可以看出, 包括5×5×1、6×6×1、7×7×1、8×8×1和9×9×1在内的5个石墨烯超晶胞的碳空位形成能分别为7.810、7.600、7.825、7.809和7.718 eV.基于非周期性石墨烯体系的DFT-PBE计算, 其碳空位形成能为7.6~7.9 eV.计算的Edef为正值, 表明在这些石墨烯超晶胞上形成碳空位缺陷是吸热过程(Ulman et al., 2014;Dai et al., 2011;Wang et al., 2012).通过计算发现, 6×6×1碳空位缺陷的Edef值最低为7.600 eV.这表明缺陷型6×6×1石墨烯比其他形式的缺陷石墨烯更易形成.
羟基化缺陷石墨烯(HO-Gdef)的Ebind计算类似公式(1), 只需将G改变为Gdef, 计算结果见表 1.由表 1计算值可见, Gdef与—OH基团之间的相互作用比G与—OH基团之间的相互作用强, 这表明缺陷型石墨烯更易被羟基化(Stankovich et al., 2006;Bagri et al., 2010;Ouyang et al., 2008;Nabil et al., 2008).基于Edef、Ebind(HO-G)和Ebind(HO-Gdef), 本研究选择HyG(6×6×1)来讨论NO和HyG之间吸附反应.
3.2 优化后HyG的电子结构特性催化剂的电子特性决定其催化活性.图 2比较了HO-G和HO-Gdef的最高占据分子轨道(Highest Occupied Molecular Orbital, HOMO)和最低未占据分子轨道(Lowest Unoccupied Molecular Orbital, LUMO).对于HO-G体系, 石墨C原子和—OH基团的O原子之间发生杂化, 石墨烯片层原子在垂直于平面方面发生4.46%弛豫.弛豫的HO-G结构, 其HOMO和LUMO主要由石墨烯的Pz原子轨道贡献.对于HO-Gdef体系, —OH基团与石墨烯的结合导致石墨烯片层原子在垂直于平面方面发生4.46%弛豫.弛豫的HO-Gdef, 其LUMO和HOMO主要由π键分子轨道贡献, 有利于电子和空穴在石墨烯上传输.
 |
| 图 2 NO与优化后的HO-G和H-Gdef之间发生在最高占据分子轨道(HOMO)和最低空分子轨道(LUMO)上可能的电子转移的示意图 Fig. 2 The schematic of possible electron transfer between NO and the optimized HO-G and H-Gdef occurring to the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) |
HO-G和HO-Gdef在与NO分子相互作用时, 可作为氧化剂, 电子将从NO分子的HOMO转移到HO-G或HO-Gdef复合材料的LUMO.根据图 2, NO分子的HOMO与HO-G复合材料的LUMO之间的能量差比NO分子的HOMO与HO-Gdef的LUMO之间的能量差小0.091 eV.此外, 比较HO-G和HO-Gdef体系中—OH基团的总电子态密度(Density of State, DOS)发现, HO-G的—OH基团在低于且靠近费米能级(0 eV)具有更多的电子密度.因此, HO-G复合材料的—OH基团的价带电子更容易激发, 化学活性将更高.
进一步分析HO-G和HO-Gdef电子得失的能力.材料获得电子时, 以电核密度变化评估材料的氧化性能.密度变化可按下式计算:
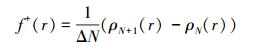
|
(3) |
式中, ρN为HyG周围空间r处的电子密度;N为HyG中的电子数量;N+1是HyG的LUMO获得一个电子形成阴离子的电子数.通过f+(r)计算获得的HO-G和HO-Gdef的密度变化分别为1.003和0.990, 这反映了HO-G比HO-Gdef更易受NO影响.这个结果与上面的DOS(图 3)和轨道分析结果一致.
 |
| 图 3 NO和—OH的DOS图 Fig. 3 DOS for NO and —OH |
在石墨烯表面上—OH基团附近的C顶位、桥位和中心位上进行NO的吸附, 获得的最稳定吸附构型如图 4所示.从图 4可见, 在NO和—OH(或石墨烯)之间不存在电子密度, 表明NO分子与HO-G(或HO-Gdef)之间是物理吸附, 仅发生范德华相互作用.HO-G和HO-Gdef与NO的N原子的距离分别为0.274 nm和0.263 nm.在NO接近HO-G和HO-Gdef表面时, NO/HO-G和NO/HO-Gdef的N—O键长比纯NO分子键长(0.116 nm)短, 分别是0.115 nm和0.115 nm.
 |
| 图 4 HO-G和HO-Gdef吸附的NO分子的稳定构型(总电荷密度) Fig. 4 Stable atomic configurations (with total charge density) for NO molecules adsorbed onto HO-G and HO-Gdef |
可通过计算NO在HO-G和HO-Gdef表面吸附的吸附能(Eads)来衡量吸附相互作用的强度, 计算公式如下:

|
(4) |
式中, Esorbate-sorbent是NO吸附在HO-G(或HO-Gdef)上的总能量(eV), Esorbent是HO-G(或HO-Gdef)的总能量(eV), Esorbate是NO分子的总能量(eV).
计算得到的NO分子的吸附能(Eads)、NO分子上的电荷总数(QNO)和NO键长(LN-O)列于表 2.从表中数据可知, NO与HO-G(或HO-Gdef)相互作用, 导致NO上电子密度减少, 因此, N—O键略微缩短.NO与HO-G(或HO-Gdef)之间相互作用, NO没有明显的杂化.通过对原子结构和吸附能的分析, 可知NO在HO-G(或HO-Gdef)上的吸附属于物理吸附, 且吸附过程为弱吸热过程;碳空位缺陷的存在不利于NO的吸附.
| 表 2 吸附能(Eads)、NO分子上的电荷数量(QNO)和N—O键长(LN—O) Table 2 The adsorption energy (Eads), charge population on NO molecule (QNO), and N—O bond length (LN—O) |
进一步研究HyG氧化NO的反应机理, 反应始态为NO在HO-G(或HO-Gdef)上的文星吸附模型(图 4).图 5给出了HO-G(或HO-Gdef)的—OH基团氧化NO的势能曲线.由于HO-G的—OH基团的DOS比HO-Gdef的—OH基团的DOS在高能级轨道分布密度更高, 因此, HO-Gdef氧化NO反应通道比HO-G氧化NO反应通道需要更多的能量, 即NO与HO-G反应生成G和NO2H的活化能Ea=0.190 eV, 而NO与HO-Gdef反应生成Gdef和NO2H的活化能Ea=2.586 eV.通常反应活化能低于0.75 eV的反应过程可在常温下发生(Wang et al., 2008).因此, 从反应势垒上来看, HO-G与NO在常温下可发生反应;然而, HO-Gdef氧化NO反应难以发生.此外, NO+HO-Gdef→ NO2H+GDEF的反应焓为1.464 eV, 反应是吸热过程, 而NO+HO-G→NO2H+G过程是放热的.热力学结果也表明, HO-G氧化NO比HO-Gdef氧化NO容易.NO与HO-G之间的相互作用体系在初始状态(IS)、过渡态(TS)和最终状态(FS)的原子电荷分布, 以及NO与HO-Gdef之间的相互作用体系在初始状态(IS)、过渡态(TS)和最终状态(FS)的原子电荷分布表明, 相互作用过程中部分电子密度从NO分子转移到—OH基团.经过电子密度重新布居, HyG可较容易地将NO氧化为NO2H.
 |
| 图 5 以eV给出的HO-G (a)和HO-Gdef (b)氧化NO的计算势能曲线(下划线标出了原子的电荷) Fig. 5 Calculated potential energy profiles given in eV for the oxidation of NO by HO-G (a) and by HO- Gdef (b) |
如上所述, HO-G氧化NO反应比HO-Gdef氧化NO容易, 但羟基化反应通常发生在有碳缺陷的石墨烯上.因此, 进一步研究HO-Gdef氧化NO的反应动力学比研究HO-G氧化NO的反应动力学更具实际意义.烟气脱硝反应通常包括高温脱硝、中温脱硝和低温脱硝, 温度范围较宽.因此, 本文研究了温度对NO和HO-Gdef反应的影响.图 6给出了NO和HO-Gdef反应与温度的关系曲线, 其中, ΔGr=GFS-GIS, ΔGα=GTS-GIS, ΔGβ =GFS-GTS(GIS、GTS、和GFS表示初始状态、过渡态和最终态的吉布斯能).根据计算得到的热力学性质, 似乎在较宽的温度范围内, 从IS到FS和从IS到TS两者都可能发生.但只有当温度高于600 K时, 从TS到FS的过程才会发生.
 |
| 图 6 NO和HO-Gdef反应与温度的关系 Fig. 6 Temperature dependence of ΔGr, ΔGα and ΔGβ for thereaction between NO and HO-Gdef |
由于NO吸附在石墨烯表面, HO-Gdef的还原遵循一步反应机理, 其中, NO直接与—OH基团相互作用, 穿越能量势垒, 转变成NO2H.所以最可能的路线如下:
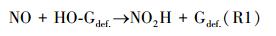
|
(5) |
式中, NO是吸附的NO分子;HO-Gdef是催化剂, —OH基团固定在石墨烯表面上.反应(R1)的净反应速率记为:
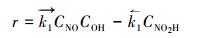
|
(6) |
式中, 
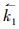
R1的正向速率常数可写为:
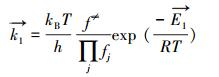
|
(7) |
式中, 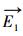
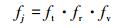
|
(8) |
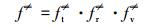
|
(9) |
式中, ft、fr和fv是反应R1中反应物的平移、旋转和振动的自由度, 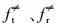
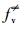
根据XPS表征, GO系统中的—OH基团含量约为10%~40%(Das et al., 2013).假设HO-Gdef的密度为1.00 g·cm-3, —OH基团为10%时, 动力学方程可以变为:

|
(10) |
用上述近似值可知, 在各种温度下HO-Gdef被烟气中NO还原的总反应速率(r)可由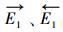
 |
| 图 7 在700 K (a)和900 K (b)下反应速率随NO和NO2H浓度(100 ~1000 mol·L-1)的变化 Fig. 7 The reaction rate, r, as a function of CNO and CNO2Hconcentration ranging from 100 to 1000 mol·L-1 under 700 K(a) and 900 K(b) |
采用DFT计算研究了烟气脱硝过程中HyG对NO的氧化机理, 并对5×5×1、6×6×1、7×7×1、8×8×1和9×9×1等各种超晶胞的HyG进行了优化, 发现6×6×1石墨烯的碳空位缺陷生成能最小为7.600 eV.随着碳缺陷形成, —OH基团牢固地结合至缺陷位点, 结合能为-4.395 eV.而无缺陷6×6×1石墨烯的结合能仅为-1.001 eV.对比研究了有碳缺陷和无碳缺陷的羟基化石墨烯HyG和HyGdef.氧化NO的反应特性, 发现NO首先物理吸附在HyG靠近—OH基团处, 然后与NO之间发生电荷重排, 越过0.190 eV势垒, NO被氧化为NO2H.NO物理吸附在HyGdef.靠近—OH基团处, 被吸附的NO与吸附表面HyG之间的电子密度发生重排后, NO和—OH相互作用发生反应, 反应越过2.596 eV的势垒, NO被氧化为NO2H.结果表明, 无缺陷HyG更容易氧化烟气中的NO.而600 K以上, HyGdef.氧化NO的反应热力学可行, 但反应只可在较高的NO浓度和很低的NO2H浓度条件下获得正的反应速率.即HyGdef.氧化NO反应高温下热力学可行, 但动力学基本不可行, HyGdef.可视为惰性载体.研究结果有助于了解石墨烯基催化剂在烟气中的反应活性及化学稳定性.
Acik M, Lee G, Mattevi C, et al. 2015. The role of oxygen during thermal reduction of graphene oxide studied by infrared absorption spectroscopy[J]. Journal of Physical Chemistry C, 115(40): 19761–19781.
|
Alaqtash N, Vasiliev I. 2009. Ab initio study of carboxylated graphene[J]. The Journal of Physical Chemistry C, 113(30): 12970–12975.
DOI:10.1021/jp902280f
|
Bagri A, Grantab R, Medhekar N V, et al. 2010. Stability and formation mechanisms of carbonyl-and hydroxyl-decorated holes in graphene oxide[J]. Journal of Physical Chemistry C, 114(28): 12053–12061.
DOI:10.1021/jp908801c
|
Castro E V, Novoselov K S, Morozov S V, et al. 2010. Electronic properties of a biased graphene bilayer[J]. Journal of Physics, 22(17): 1060–1065.
|
Dai X Q, Zhao J H, Xie M H, et al. 2011. First-principle study of magnetism induced by vacancies in graphene[J]. European Physical Journal B, 80(3): 343–349.
DOI:10.1140/epjb/e2011-10955-x
|
Dreyer D R, Jarvis K A, Ferreira P J, et al. 2010. Graphite oxide as a dehydrative polymerization catalyst:A one-step synthesis of carbon-reinforced poly(phenylene methylene) composites[J]. Macromolecules, 44(19): 7659–7667.
|
Das S, Singh S, Singh V, et al. 2013. Oxygenated functional group density on graphene oxide:its effect on cell toxicity[J]. Particle & Particle Systems Characterization, 30(2): 148–157.
|
El-Barbary A A, Telling R H, Ewels C P, et al. 2003. Structure and energetics of the vacancy in graphite[J]. Physical Review B Condensed Matter, 68(14): 144107/1–144107/7.
|
Gao H, Liu Z. 2017. Dft study of no adsorption on pristine graphene[J]. RSC Advances, 7(22): 13082–13091.
DOI:10.1039/C6RA27137E
|
Han S, Wu D, Li S, et al. 2014. Porous graphene materials for advanced electrochemical energy storage and conversion devices[J]. Advanced Materials, 26(6): 849–864.
|
Halgren T A, Lipscomb W N. 1977. The synchronous-transit method for determining reaction pathways and locating molecular transition states[J]. Chemical Physics Letters, 49(2): 225–232.
DOI:10.1016/0009-2614(77)80574-5
|
Hernandez Y, Nicolosi V, Lotya M, et al. 2008. High yield production of graphene by liquid phase exfoliation of graphite[J]. Nature Nanotechnology, 3(9): 563–568.
|
Heyrovska R. 2008. Atomic structures of graphene, benzene and methane with bond lengths as sums of the single, double and resonance bond radii of carbon[J]. Chemical Physics: arXiv:0804.4086.
|
Jr W S, Offeman R E. 1958. Preparation of graphitic oxide[J]. Journal of the American Chemical Society, 80(6): 1339–1339.
DOI:10.1021/ja01539a017
|
Krishna R, Titus E, Okhay O, et al. 2014. Rapid electrochemical synthesis of hydrogenated graphene oxide using Ni nanoparticles[J]. International Journal of Electrochemical Science, 9(7): 4054–4069.
|
Kyotani T, Tomita A. 1999. Analysis of the reaction of carbon with NO/N2O using ab initio molecular orbital theory[J]. Journal of Physical Chemistry B, 103(17): 275–278.
|
Lerf A, He H, Forster M, et al. 1999. Structure of graphite oxide revisited[J]. The Journal of Physical Chemistry B, 102(23): 4477–4482.
|
Li J H, Lin C F, Qin W, et al. 2016. Synergetic effect of mercury adsorption on the catalytic decomposition of CO over perfect and reduced Fe2O3[001] surface[J]. Acta Physico-Chimica Sinica, 32(11): 2717–2723.
|
梁志永, 王建业, 覃吴, 等. 2017. Fe2O3(1-12)作用下CO化学链燃烧反应机理研究[J]. 环境科学学报, 2017, 37(5): 1826–1834.
|
Liu J, Durstock M, Dai L. 2014. Graphene oxide derivatives as hole-and electron-extraction layers for high-performance polymer solar cells[J]. Energy & Environmental Science, 7(4): 1297–1306.
|
Mochida I, Kisamori S, Hironaka M, et al. 1994. Oxidation of NO into NO2 over active carbon fibers[J]. Energy & Fuels, 8(6): 1341–1344.
|
Ouyang F, Huang B, Li Z, et al. 2008. Chemical functionalization of graphene nanoribbons by carboxyl groups on Stone-Wales defects[J]. The Journal of Physical Chemistry C, 112(31): 12003–12007.
DOI:10.1021/jp710547x
|
Park S, Lee K S, Bozoklu G, et al. 2008. Graphene oxide papers modified by divalent ions-enhancing mechanical properties via chemical cross-linking[J]. ACS Nano, 2(3): 572–578.
DOI:10.1021/nn700349a
|
Payne M C, Teter M P, Allan D C, et al. 1992. Iterative minimization techniques for ab initio total-energy calculations:Molecular dynamics and conjugate gradients[J]. Reviews of Modern Physics, 64(4): 1045–1097.
DOI:10.1103/RevModPhys.64.1045
|
Pei C C, Lo K K S, Leung W F. 2017. Titanium-zinc-bismuth oxides-Graphene composite nanofibers as high-performance photocatalyst for gas purification[J]. Separation & Purification Technology, 184: 205–212.
|
Perdew J P, Chevary J A, Vosko S H, et al. 1992. Atoms, molecules, solids, and surfaces:Applications of the generalized gradient approximation for exchange and correlation[J]. Physical Review B, 46(11): 6671–6678.
|
Poh H L, Sofer Z, et al. 2015. Hydroboration of graphene oxide:Towards stoichiometric graphol and hydroxygraphane[J]. Chemistry-A European Journal, 21(22): 8130–8136.
DOI:10.1002/chem.201406168
|
Qin W, Lin C F, Long D T, et al. 2015. Reaction activity and deep reduction reaction mechanism of a high index iron oxide surface in chemical looping combustion[J]. Acta Physico-Chimica Sinica, 31(4): 667–675.
|
Robinson J T, Perkins F K, Snow E S, et al. 2008. Reduced graphene oxide molecular sensors[J]. Nano Letters, 8(10): 3137–3140.
DOI:10.1021/nl8013007
|
Robinson J T, Zalalutdinov M, Baldwin J W, et al. 2008. Wafer-scale reduced graphene oxide films for nanomechanical devices[J]. Nano Letters, 8(10): 3441.
DOI:10.1021/nl8023092
|
Schniepp H C, Li J L, Mcallister M J, et al. 2006. Functionalized single graphene sheets derived from splitting graphite oxide[J]. The Journal of Physical Chemistry B, 110(17): 8535–8539.
DOI:10.1021/jp060936f
|
Stankovich S, Dikin D A, Dommett G H B, et al. 2006. Graphene-based composite materials[J]. Nature, 442(7100): 282–286.
|
Szabó T, Berkesi O, Forgó P, et al. 2006. Evolution of surface functional groups in a series of progressively oxidized graphite oxides[J]. Chemistry of Materials, 18(11): 2740–2749.
DOI:10.1021/cm060258+
|
Taherian F, Marcon V, van der Vegt N F A, et al. 2013. What is the contact angle of water on graphene[J]. Langmuir, 29(5): 1457–1465.
DOI:10.1021/la304645w
|
Telling R H, Ewels C P, Ahlam A, et al. 2003. Wigner defects bridge the graphite gap[J]. Nature Materials, 2(5): 333–337.
DOI:10.1038/nmat876
|
Ulman K, Narasimhan S. 2014. Point defects in twisted bilayer graphene:A density functional theory study[J]. Physical Review B, 89(24): 2495–2502.
|
Vanderbilt D. 1990. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Physical Review B, 41(11): 7892–7895.
DOI:10.1103/PhysRevB.41.7892
|
Wallace P R. 1947. The band theory of graphite[J]. Physical Review, 71(9): 622–634.
DOI:10.1103/PhysRev.71.622
|
Wang Z, Yan S, Sun Y, et al. 2017. Bi metal sphere/graphene oxide nanohybrids with enhanced direct plasmonic photocatalysis[J]. Applied Catalysis B:Environmental, 214: 148–157.
DOI:10.1016/j.apcatb.2017.05.040
|
Wang B, Pantelides S T. 2012. Magnetic moment of a single vacancy in graphene and semiconducting nanoribbons[J]. Physical Review B, 86(16): 5505–5511.
|
Wang H F, Liu Z P. 2008. Comprehensive mechanism and structure-sensitivity of ethanol oxidation on platinum:New transition-state searching method for resolving the complex reaction network[J]. Journal of the American Chemical Society, 130(33): 10996–11004.
|
Wei N, Lv C, Xu Z. 2014. Wetting of graphene oxide:A molecular dynamics study[J]. Langmuir, 30(12): 3572–3578.
DOI:10.1021/la500513x
|
White J A, Bird D M. 1994. Implementation of gradient-corrected exchange-correlation potentials in Car-Parrinello total-energy calculations[J]. Physical Review B, 50(7): 4954–4957.
DOI:10.1103/PhysRevB.50.4954
|