 2019, Vol. 39
2019, Vol. 39



2. 中国市政工程中南设计研究总院有限公司, 武汉 430010
2. Central and Southern China Municipal Engineering Design & Research Institute Co., Ltd, Wuhan 430010
Fe2+是一种良好的活化过硫酸盐(PS)的试剂, 有活化速率高、成本低、对环境友好等许多优点, 具有非常广泛的应用前景(Liu et al., 2018;Rastogi et al., 2009), 但是Fe2+活化PS也有非常明显的缺点, 比如其持续活化能力不足, 因此在活化PS时需要投加大量的Fe2+, 而Fe2+与PS反应后生成的Fe3+很容易水解而生成铁泥, 降低活化效率, 并且其适用pH范围比较窄(Xu et al., 2010).Fe2+活化PS的这些缺点也限制了其应用, 因此需要考虑如何提高Fe2+活化PS的效率.一般可以通过加入络合剂、还原剂、零价铁(Fe0)等方式提高其活化效率, 其中, 加入还原剂对Fe2+/PS体系的强化作用最强.
羟胺(HA)是一种强还原剂, 在使用邻二氮菲分光光度法测定水中的总铁时, 就是利用HA将水中的Fe3+还原成Fe2+(Harvey et al., 1955).已经有文献报道, HA可以促进Fenton体系和Fe2+/PMS以及Fe2+/PS体系中Fe3+转化为Fe2+, 强化其对目标污染物的降解效果(Chen et al., 2011;邹景, 2016).因此可以考虑向Fe2+/PS体系中加入HA促进Fe3+还原成Fe2+从而强化其对ACT的降解效果.一般使用较多的含HA的化合物为盐酸羟胺(NH2OH·HCl)和硫酸羟胺((NH2OH)2·H2SO4), 考虑到Cl-对Fe2+/PS体系降解ACT有影响, 选择硫酸羟胺投加到Fe2+/PS体系.
现以ACT为目标污染物, 研究HA强化Fe2+/PS体系的去除效果, 考察Fe2+、PS、HA投加浓度以及pH值对ACT降解效果的影响, 并分析其反应机理, 并且与一些常见的还原剂强化Fe2+/PS体系降解ACT的效果进行对比.
2 材料与方法(Materials and methods) 2.1 实验材料实验所用过硫酸盐、硫酸亚铁、碘化钾、醋酸、邻菲罗啉、硫酸羟胺、亚硝酸钠、硫代硫酸钠均为分析纯, 购于国药集团化学试剂有限公司, 对乙酰氨基酚、5, 5-二甲基-1-氧化吡咯啉、N, N-二乙基对苯二胺硫酸盐均为分析纯, 购于Aladdin公司, 甲醇为色谱纯, 购于Tedia公司, 所有试剂的溶解和配制均用超纯水.
实验用到的主要仪器有高效液相色谱仪, LC-16型, Shimadzu公司生产; pH计, PHS-3C型, 上海仪电科学仪器公司生产; 紫外可见分光光度计, UV-3100型, 上海美普达公司生产; 电子顺磁共振波谱仪, EMX nano型, Bruker Biospin公司生产; 超纯水仪, PCDX-J型, 成都品成科技公司生产; 总有机碳测定仪, Multi N/C 3100型, Jena公司生产.
2.2 实验方法本实验反应器采用双层夹套烧杯, 恒温水浴槽的进出水口与夹套烧杯相连接, 提供循环恒温水, 维持反应液恒温.Fe2+/PS体系降解ACT的实验步骤如下:反应开始之前计算出要加入的超纯水以及各试剂的体积, 先将超纯水加入反应器中, 开启磁力搅拌器和恒温水浴槽, 设定水浴槽温度为20 ℃, 将温度计放入反应器内, 待温度维持在(20±1) ℃时, 加入ACT和FeSO4, 迅速用0.1 mol·L-1的HClO4或NaOH调节pH值设定值, 随后加入Na2S2O8进行反应, 同时开始计时.分别在1、2、3、5、10、20、30 min取样1 mL, 然后迅速加0.1 mL甲醇淬灭, 经0.22 μm滤膜过滤后采用高效液相色谱仪分析ACT浓度.
2.3 分析方法对乙酰氨基酚的测定采用高效液相色谱仪(HPLC).分析条件如下:色谱柱型号XDB-C18(5 μm, 4.6 mm×150 mm), 流动相为甲醇-0.1%乙酸=30:70, 流速为0.8 L·min-1, 柱温30 ℃, UV检测器(SPD-16, Shimadzu), 检测波长为243 nm, 进样体积20 μL.
实验过程中Fe2+的浓度采用邻菲罗啉分光光度法测定(Harvey et al., 1955), 其原理是在pH 3.0~9.0的条件下, Fe2+与邻菲罗啉生成稳定的橙红色络合物, 其吸光度值与Fe2+的浓度成正比.
对于Fe2+/PS体系, 反应过程中PS浓度采用碘量法测定(Liang et al., 2008), 其原理是利用PS将I-氧化生成I2, 测定其中I2的吸光度, 从而确定PS浓度.
本实验中TOC采用德国Jena公司生产的Multi N/C 3100总有机碳测定仪进行测定.由于采用甲醇作为淬灭剂会对TOC的测定产生干扰, 因此在测定TOC时采用NaNO2作为淬灭剂.
Fe2+/PS/HA体系中产生的SO4·-与HO·用5, 5-二甲基-1-氧化吡咯啉(DMPO)捕获, 采用电子顺磁共振波谱仪(EPR)进行检测.EPR设置磁场强度的范围3360~3460 G, 扫描次数3次.
3 结果与讨论(Results and discussion) 3.1 羟胺强化Fe2+/PS体系与原体系的比较为了探究HA强化Fe2+/PS体系对ACT的降解效果, 对比Fe2+/PS、HA/PS、Fe2+/PS/HA 3种体系对ACT的降解效果, 其结果如图 1所示.可以看到, Fe2+/PS对ACT的降解效果不理想, 30 min内ACT的去除率只有13%, 是由于Fe2+浓度较低, 不能通过活化PS产生足够的自由基来降解ACT.HA/PS体系对ACT的降解效果很弱, 30 min内ACT的去除率只有8%, 表明HA不能活化PS降解ACT.而Fe2+/PS/HA体系却能够有效的降解ACT, 30 min内ACT的去除率达到了90%, 表明HA能够极大强化Fe2+/PS对ACT的降解, 推测可能是HA促进了体系中Fe3+ /Fe2+的循环, 导致有更多的PS被活化产生自由基来降解ACT.
 |
| 图 1 Fe2+/PS、HA/PS、Fe2+/PS/ HA体系对ACT的降解效果 (实验条件:[ACT]0=0.05 mmol·L-1, pH=3.0, [Fe2+]0=0.05 mmol·L-1, [PS]0=0.8 mmol·L-1, [HA]0=0.5 mmol·L-1) Fig. 1 Degradation of ACT in different reaction systems |
为了验证以上猜测, 检测了不同体系中Fe2+和PS的浓度变化情况, 其结果如图 2所示.可以看到, 在Fe2+/PS体系中, Fe2+浓度在1 min内有97%被氧化成Fe3+, 此后体系中Fe2+浓度一直维持在5%以下, 这表明Fe2+能被PS快速氧化成Fe3+并且无法还原重生成Fe2+, 因此Fe2+/PS体系对ACT的降解效果较差.Fe2+/PS体系中PS浓度在30 min内只减少了9%, 这也验证了该反应没有足够的Fe2+活化PS产生自由基来降解ACT.而在Fe2+/PS/HA体系中, Fe2+浓度在1 min内有37%被氧化成Fe3+, 此后Fe2+浓度以比较缓慢的速度下降, 30 min内有90%的Fe2+浓度被氧化成Fe3+, 这说明HA能够减缓Fe2+/PS体系中Fe2+浓度的变化速率.这有可能通过两种途径实现, 一是通过减缓Fe2+与PS的反应速率, 二是通过将体系中Fe3+还原成Fe2+.比较Fe2+/PS/HA、HA/PS、Fe2+/PS体系中PS浓度的变化情况可以看到, HA/PS体系中PS浓度变化情况与Fe2+/PS体系类似, 30 min内只有不到10%的PS消耗, 这说明HA与PS反应比较慢, 而在Fe2+/PS/ HA体系中, 30 min内有54%的PS消耗, 这说明HA是通过促进Fe3+还原成Fe2+而提高了PS的活化程度, 从而产生更多的自由基来降解ACT.根据文献资料, HA还原Fe3+的过程主要有以下反应(Butler et al., 1986; Bengtsson et al., 2002; Johnson et al., 2003; Chen et al., 2011):
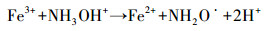
|
(1) |

|
(2) |
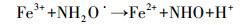
|
(3) |
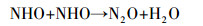
|
(4) |

|
(5) |
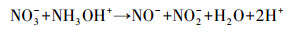
|
(6) |
 |
| 图 2 不同体系中Fe2+(a)、PS(b)浓度变化 (实验条件:[ACT]0=0.05 mmol·L-1, pH=3.0, [Fe2+]0=0.05 mmol·L-1, [PS]0=0.8 mmol·L-1, [HA]0=0.5 mmol·L-1) Fig. 2 The concentration of Fe2+(a), PMS(b) in different reaction systems |
虽然HA具有一定的危害(Johnson et al., 2003), 但是有研究表明, 当HA浓度高于Fe3+时, HA与Fe3+反应的主要产物是无害的N2, 且当PS浓度高于HA时, HA基本可以完全分解(Bengtsson et al., 2002;邹景, 2016).本实验中Fe2+、PS、HA三者的浓度大小为PS>HA>Fe2+, 因此认为加入HA不会引入新的有毒物质.
3.2 羟胺投加浓度的影响考察HA投加浓度对Fe2+/PS/HA体系降解ACT的影响, 其结果如图 3所示.从图中可以看到, 当HA浓度在0~0.5 mmol·L-1时, 随着HA投加浓度的增加, ACT的降解率也随之增加.例如, 当HA投加浓度为0.025 mmol·L-1时, 30 min内ACT的降解率为55%, 而当HA投加浓度为0.5 mmol·L-1时, 30 min内ACT的降解率达到了90%.其原因主要是因为增加HA的浓度可以提高Fe3+转化为Fe2+的转化速率和转化的总量, 导致有更多的PS被活化产生SO4·-和HO·来降解ACT.同时也应当注意到, 当HA浓度超过0.5 mmol·L-1时, 将不能进一步促进ACT的降解, 例如, 当HA的投加浓度为1 mmol·L-1时, 30 min内ACT的降解率为89%, 与HA的投加浓度为0.5 mmol·L-1时ACT的降解率相近.而当HA的投加浓度进一步增加到2 mmol·L-1时, 30 min内ACT的降解率反而略微下降至84%, 这是因为HA也会与SO4·-和HO·反应(Neta et al., 1988; Buxton et al., 1988), 当HA的浓度过高时, 会与ACT竞争体系中产生的SO4·-和HO·, 因此在采用HA强化Fe2+/PS降解污染物时要选择合适的HA投加浓度, 避免因HA浓度过高造成对目标污染物的降解效果不理想.
 |
| 图 3 HA投加浓度对Fe2+/PS/HA体系降解ACT的影响 (实验条件:[ACT]0=0.05 mmol·L-1, pH=3.0, [Fe2+]0=0.05 mmol·L-1, [PS]0=0.8 mmol·L-1) Fig. 3 Effect of HA dosage of the Fe2+/PS/HA process for the degradation of ACT |
图 4描述了Fe2+投加浓度对Fe2+/PS/HA体系降解ACT的影响.可以看到当Fe2+浓度在0~0.05 mmol·L-1时, 提高Fe2+的投加浓度可以提高ACT的降解效果, 但是当Fe2+浓度高于0.1 mmol·L-1时, 提高Fe2+的投加浓度将会对ACT的降解效果产生抑制作用.非常低的Fe2+浓度就能对ACT有较好的降解效果, 例如, 当Fe2+浓度为0.005 mmol·L-1时, 30 min内ACT的降解率达到了58%, 这是因为HA促进了Fe3+/Fe2+循环, 提高了PS的活化效果, 所以对ACT的降解有一定效果.当Fe2+的投加浓度增加到0.05 mmol·L-1时, 30 min内ACT的降解率达到了90%, 这是因为提高Fe2+的浓度可以促进更多的PS被活化产生自由基, 所以ACT的降解率也随之提高.然而, 当Fe2+的投加浓度提高至0.1 mmol·L-1时, ACT的降解效果并没有提高, 而当Fe2+的投加浓度提高至0.4 mmol·L-1时, ACT的降解效果反而降低, 这是由于Fe2+能与体系中的SO4·-和HO·反应, 并且其反应速率非常高(Kusic et al., 2011; Bu et al., 2016), 因此当Fe2+浓度增加到一定程度后会对自由基产生淬灭作用, 从而导致ACT的降解效果受到抑制.此外, 可以看到在不同Fe2+投加浓度下, Fe2+/PS/HA体系对ACT的降解并没有明显的快反应和慢反应阶段, 这也是因为HA促进了体系中Fe3+还原成Fe2+.
 |
| 图 4 Fe2+投加浓度对Fe2+/PS/HA体系降解ACT的影响 (实验条件:[ACT]0=0.05 mmol·L-1, pH=3.0, [PS]0=0.8 mmol·L-1, [HA]0=0.5 mmol·L-1) Fig. 4 Effect of Fe2+ dosage of the Fe2+/PS/HA process for the degradation of ACT |
PS浓度对目标污染物在活化过硫酸盐体系中的降解有重要影响, 因为PS是各种自由基的直接来源, PS浓度过低, 将不能产生足够的自由基降解污染物, 而PS浓度过高, 将会造成PS的浪费.考察不同PS投加浓度对Fe2+/PS/HA体系降解ACT的影响, 其结果如图 5所示.可以看到, 随着PS投加浓度的提高, Fe2+/PS/HA体系对ACT的降解率也随之升高, 当PS投加浓度为0.2 mmol·L-1时, 30 min内ACT的降解率为36%, 而当PS浓度升高至1.2 mmol·L-1时, 30 min内ACT的降解率达到了97%, 这是因为提高体系中PS的浓度导致更多的PS被活化产生SO4·-和HO·来降解ACT.而当PS浓度进一步增加至1.6 mmol·L-1时, 30 min内ACT已经被完全降解.这与以前得到的结果有所不同, 在Fe2+/PS体系中, 增加PS投加浓度并不能完全降解ACT, 而在Fe2+/PS/HA体系中增加PS投加浓度则可以完全降解ACT, 这表明Fe2+/PS/HA体系相比于Fe2+/PS体系有优势, 进一步说明了HA对Fe2+/PS体系的强化作用.
 |
| 图 5 PS投加浓度对Fe2+/PS/ HA体系降解ACT的影响 (实验条件:[ACT]0=0.05 mmol·L-1, pH=3.0, [Fe2+]0=0.05 mmol·L-1, [HA]0=0.5 mmol·L-1) Fig. 5 Effect of PS dosage of the Fe2+/PS/ HA process for the degradation of ACT |
pH能够通过多个方面影响目标污染物在Fe2+/PS体系中的降解效果, 以前的研究表明, pH主要通过影响Fe2+的形态而影响其与氧化剂的反应速率(Wang et al., 2011), 从而影响ACT的降解效果, 此外, pH还会影响HA在水中的存在形态(Robinson et al., 1961; Hughes et al., 1971), 如图 6所示, 而不同形态的HA与自由基的反应速率有很大的差别(Buxton et al., 1988; Neta et al., 1988), 因此需要考察不同pH条件下Fe2+/PS/HA体系对ACT的降解效果.pH对Fe2+/PS/HA体系降解ACT的影响如图 7所示.
 |
| 图 6 不同pH条件下羟胺在水中的形态分布 Fig. 6 Morphological distribution of hydroxylamine in water under different pH conditions |
 |
| 图 7 pH值对Fe2+/PS/HA体系降解ACT的影响 (实验条件:[ACT]0=0.05 mmol·L-1, [Fe2+]0=0.05 mmol·L-1, [PS]0=0.8 mmol·L-1, [HA]0=0.5 mmol·L-1) Fig. 7 Effect of pH of the Fe2+/PS/HA process for the degradation of ACT |
从整体上看, 随着pH的增加, Fe2+/PS/HA体系对ACT的降解效果逐渐降低.pH=3时, 30 min内ACT的降解率为90%, 在pH=5条件下ACT的降解率与pH=3时几乎没有变化, pH>4时会阻碍Fe2+与PS的反应速率, 从而降低目标污染物的降解效果(Xu et al., 2010).监测Fe2+/PS/HA体系的pH变化情况(图 8)发现, 在初始pH=3条件下, 体系的pH几乎没有变化, 初始pH=5时, 体系的pH在3 min内迅速下降至3左右, 因此导致初始pH=5条件下ACT的降解率与pH=3条件下几乎没有区别.当初始pH=7时, Fe2+/PS/HA体系对ACT的降解率在30 min内下降至74%.可以看到, 初始pH=7条件下体系的pH在30 min内也会逐渐下降至3左右, 而在反应的前5 min内, 体系的pH>4, 导致Fe2+与PS的反应速率降低, 这可能是导致ACT的降解效果降低的重要原因.图 7的结果也表明, 初始pH=7与初始pH=3条件下ACT降解效果的差别主要也是体现在前5 min, 此后两者的降解速率接近相同.当初始pH升高至9时, ACT的降解效果极差, 30 min内的降解率只有12%, 而体系的pH在30 min内也只能下降至6左右, 在此pH下Fe2+与PS的反应速率较低, 是导致ACT的降解效果较低的原因.另外, pH>6时HA主要以NH2OH的形式存在, 而NH2OH与SO4·-和HO·的反应速率远高于NH3OH+, 这也是导致ACT的降解效果降低的一个重要因素.
 |
| 图 8 不同初始pH条件下Fe2+/PS/HA体系的pH随时间的变化 (实验条件:[ACT]0=0.05 mmol·L-1, [Fe2+]0=0.05 mmol·L-1, [PS]0=0.8 mmol·L-1, [HA]0=0.5 mmol·L-1) Fig. 8 Changes of pH with time of Fe2+/PS/HA system under different initial pH conditions |
为了验证Fe2+/PS/HA体系降解ACT过程中的自由基类型, 采用电子顺磁共振波谱仪(Electron Paramagnetic Resonance, EPR)检测反应过程中的自由基, 利用5, 5-二甲基-1-氧化吡咯啉(DMPO)作为自由基捕获剂, DMPO可以与SO4·-和HO·结合形成特定的自旋加成产物, 利用其产物特定的特征峰以及超精细耦合常数鉴别SO4·-和HO· (Jawad et al., 2018).其中, DMPO与HO·结合形成峰高为1:2:2:1的四重峰, 超精细耦合常数为a(N)=a(H)=14.7 G, DMPO与SO4·-结合形成峰高为1:1:1:1:1:1的六重峰, 超精细耦合常数为a(N)=13.3 G, a(H)=9.5 G, a(H)=1.46 G, a(H)=0.77 G(Zhu et al., 2016).
图 9显示了EPR检测Fe2+/PS/ HA体系中自由基的结果.从EPR谱图中可以看到有DMPO-OH和DMPO-SO4的特征峰出现, 证明了Fe2+/PS/ HA体系中的主要自由基是SO4·-和HO·.其中DMPO-OH的特征峰高度比DMPO-SO4要高得多, 这可能是因为DMPO-SO4在水溶液中容易发生水解反应生成DMPO-OH(Timmins et al., 1999).
 |
| 图 9 Fe2+/PS/HA体系自由基的EPR谱图 (实验条件:[Fe2+]0=1.0 mmol·L-1, [PS]0=16 mmol·L-1, pH=3.0, [HA]0=10 mmol·L-1, [DMPO]0=100 mmol·L-1) Fig. 9 EPR spectra of Fe2+/PS/HA system |
由前文分析可知, HA作为一种还原剂, 主要通过促进Fe2+/PS体系中Fe3+转化为Fe2+来强化Fe2+/PS体系对ACT的降解效果, 那么其它还原剂是否也能够强化Fe2+/PS体系对ACT的降解效果.为了探究这个问题, 选取了常见的3种还原剂:亚硫酸钠(Na2SO3)、亚硝酸钠(NaNO2)和硫代硫酸钠(Na2S2O3), 考察其对Fe2+/PS体系降解ACT的影响, 结果如图 10所示.可以看到, 3种还原剂对Fe2+/PS体系降解ACT都有一定的强化效果, 表明这3种还原剂都能够促进Fe3+转化为Fe2+, 但是过高的Na2SO3和Na2S2O3浓度反而会抑制ACT的降解.当Fe2+和PS浓度分别为0.05 mmol·L-1和0.8 mmol·L-1时, Na2SO3和Na2S2O3的最佳投加浓度分别为1 mmol·L-1和0.2 mmol·L-1, 对应的ACT的最佳降解效果分别为56%和71%, 此后继续增加Na2SO3和Na2S2O3浓度会抑制ACT降解, 当Na2SO3和Na2S2O3浓度升高至2 mmol·L-1时, 30 min内ACT的降解率分别降低至12%和28%.NaNO2浓度在0~2 mmol·L-1时, 增加NaNO2浓度可以提高Fe2+/PS体系对ACT的降解效果, 当NaNO2浓度为2 mmol·L-1时, 30 min内ACT的降解效果为52%.同时可以看出, Na2SO3、NaNO2和Na2S2O3等还原剂对Fe2+/PS体系的强化效果均低于HA, 这是因为当pH=3时HA(主要以NH3OH+形式存在)与SO4·-和HO·的反应速率远小于Na2SO3、NaNO2和Na2S2O3等还原剂(Neta et al., 1988; Buxton et al., 1988).
 |
| 图 10 常见的3种还原剂对Fe2+/PS体系降解ACT的影响 (实验条件:pH=3.0, [ACT]0=0.05 mmol·L-1, [Fe2+]0=0.05 mmol·L-1, [PS]0=0.8 mmol·L-1) Fig. 10 Effect of three common reducing agents on degradation of ACT in Fe2+/PS system |
1) HA能够通过加快三价铁还原为二价铁过程极大强化Fe2+/PS体系对ACT的降解效果.当Fe2+、PS和HA的投加浓度分别为0.05、0.8和0.5 mmol·L-1时, Fe2+/PS/HA体系对ACT的降解效果达到90%.
2) 适量增加Fe2+或HA浓度可以提高ACT降解率, 但是过高浓度的Fe2+和HA会抑制ACT的降解.ACT的降解率随着PS浓度升高而提升, 随着pH的升高而降低.
3) EPR实验表明Fe2+/PS/HA体系中主要的自由基是SO4·-和HO·.
4) Na2SO3、NaNO2和Na2S2O3等常见的还原剂均能够强化Fe2+/PS体系对ACT的降解效果, 但是过高浓度的Na2SO3和Na2S2O3会抑制ACT的降解, 且Na2SO3、NaNO2和Na2S2O3等还原剂对Fe2+/PS体系的强化效果均比HA低.
Bengtsson G, Fronæus S, Bengtsson-Kloo L. 2002. The kinetics and mechanism of oxidation of hydroxylamine by iron (Ⅲ)[J]. Journal of the Chemical Society, Dalton Transactions(12): 2548–2552.
DOI:10.1039/b201602h
|
Bu L, Shi Z, Zhou S. 2016. Modeling of Fe(Ⅱ)-activated persulfate oxidation using atrazine as a target contaminant[J]. Separation and Purification Technology, 169: 59–65.
DOI:10.1016/j.seppur.2016.05.037
|
Butler J H, Gordon L I. 1986. Rates of nitrous oxide production in the oxidation of hydroxylamine by iron (Ⅲ)[J]. Inorganic Chemistry, 25(25): 4573–4577.
DOI:10.1021/ic00245a024
|
Buxton G V, Greenstock C L, Helman W P, et al. 1988. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O- in aqueous solution[J]. Journal of physical and chemical reference data, 17(2): 513–886.
DOI:10.1063/1.555805
|
Chen L, Ma J, Li X, et al. 2011. Strong enhancement on Fenton oxidation by addition of hydroxylamine to accelerate the ferric and ferrous iron cycles[J]. Environmental Science & Technology, 45(9): 3925–3930.
|
Harvey Jr A E, Smart J A, Amis E S. 1955. Simultaneous spectrophotometric determination of iron (Ⅱ) and total iron with 1, 10-phenanthroline[J]. Analytical Chemistry, 27(1): 26–29.
|
Hughes M N, Nicklin H G, Shrimanker K. 1971. Autoxidation of hydroxylamine in alkaline solutions. Part Ⅱ. Kinetics. The acid dissociation constant of hydroxylamine[J]. Journal of the Chemical Society A:Inorganic, Physical, Theoretical: 3485–3487.
DOI:10.1039/j19710003485
|
Jawad A, Lang J, Liao Z, et al. 2018. Activation of persulfate by CuOx@Co-LDH:A novel heterogeneous system for contaminant degradation with broad pH window and controlled leaching[J]. Chemical Engineering Journal, 335: 548–559.
DOI:10.1016/j.cej.2017.10.097
|
Johnson M D, Hornstein B J. 2003. The kinetics and mechanism of the ferrate (Ⅵ) oxidation of hydroxylamines[J]. Inorganic Chemistry, 42(21): 6923–6928.
DOI:10.1021/ic020705x
|
Kusic H, Peternel I, Ukic S, et al. 2011. Modeling of iron activated persulfate oxidation treating reactive azo dye in water matrix[J]. Chemical Engineering Journal, 172(1): 109–121.
DOI:10.1016/j.cej.2011.05.076
|
Liang C, Huang C, Mohanty N, et al. 2008. A rapid spectrophotometric determination of persulfate anion in ISCO[J]. Chemosphere, 73(9): 1540–1543.
DOI:10.1016/j.chemosphere.2008.08.043
|
Liu Y, Lang J, Wang T, et al. 2018. Enhanced degradation of isoproturon in soil through persulfate activation by Fe-based layered double hydroxide:different reactive species comparing with activation by homogenous Fe(Ⅱ)[J]. Environ Sci Pollut Res Int, 25(26): 26394–26404.
DOI:10.1007/s11356-018-2637-3
|
Neta P, Huie R E, Ross A B. 1988. Rate constants for reactions of inorganic radicals in aqueous solution[J]. Journal of Physical and Chemical Reference Data, 17(3): 1027–1284.
DOI:10.1063/1.555808
|
Rastogi A, Al-Abed S R, Dionysiou D D. 2009. Effect of inorganic, synthetic and naturally occurring chelating agents on Fe (Ⅱ) mediated advanced oxidation of chlorophenols[J]. Water Research, 43(3): 684–694.
DOI:10.1016/j.watres.2008.10.045
|
Robinson R A, Bower V E. 1961. The ionization constant of hydroxylamine[J]. The Journal of Physical Chemistry, 65(7): 1279–1280.
DOI:10.1021/j100825a508
|
Timmins G S, Liu K J, Bechara E J, et al. 1999. Trapping of free radicals with direct in vivo EPR detection:a comparison of 5, 5-dimethyl-1-pyrroline-N-oxide and 5-diethoxyphosphoryl-5-methyl-1-pyrroline-N-oxide as spin traps for HO and SO4·-[J]. Free Radical Biology and Medicine, 27(3/4): 329–333.
|
Wang Y R, Chu W. 2011. Degradation of a xanthene dye by Fe (Ⅱ)-mediated activation of Oxone process[J]. Journal of Hazardous Materials, 186(2/3): 1455–1461.
|
Xu X, Li X. 2010. Degradation of azo dye Orange G in aqueous solutions by persulfate with ferrous ion[J]. Separation and Purification Technology, 72(1): 105–111.
DOI:10.1016/j.seppur.2010.01.012
|
Zhu C, Fang G, Dionysiou D D, et al. 2016. Efficient transformation of DDTs with persulfate activation by zero-valent iron nanoparticles:a mechanistic study[J]. Journal of Hazardous Materials, 316: 232–241.
DOI:10.1016/j.jhazmat.2016.05.040
|
邹景. 2016.羟胺对Fe2+/过硫酸盐体系的强化效能与机理研究[D].哈尔滨: 哈尔滨工业大学
|