 2019, Vol. 39
2019, Vol. 39



2. 国家环境保护有机化工废水处理与资源化工程技术中心, 南京 210046;
3. 南京大学常高新国际环保产业技术研究院, 常州 213125
2. State Environmental Protection Engineering Center for Organic Chemical Wastewater Treatment and Resource Reuse, Nanjing 210046;
3. Nanjing University-International Research Institute of Environmental Industries, Changzhou 213125
近年来, 以药品和个人护理品(Pharmaceuticals and personal care products, PPCPs)为代表的新型有机污染物备受关注(Nakada et al., 2007), 污水处理厂污水的排放是导致PPCPs进入自然水体的主要途径之一(Nikolaou et al., 2007;周海东等, 2007).尽管污水处理厂生化出水中这类污染物质的残留浓度不高(ng·L-1~μg·L-1), 但由于PPCPs能产生内分泌干扰效应、易造成耐药微生物的形成和传播等特性, 其对水生生物和人类都有较为严重的危害.因此, PPCPs的深度去除在国际上受到广泛关注(宋存义等, 2009;Michael et al., 2013; Rivera-Utrilla et al., 2013; Ribeiro et al., 2015).
常规水处理技术无法有效去除痕量的PPCPs, 臭氧工艺因具有效果可观、二次污染小等优势, 尤其适合用于生化尾水的深度处理(Audenaert et al., 2013).生化尾水中有机物组成十分复杂, 污水生化出水有机物(EfOM)往往会显著影响PPCPs的臭氧降解效率, 研究发现在真实二级出水体系中, DOC含量的不同会直接影响不同性质的PPCPs去除率和降解速率(Yao et al., 2016;Wang et al., 2018; Yao et al., 2018), 其原因可能为水中EfOM影响了O3对PPCPs的降解机制, 进而影响了处理效果.例如具有富电子芳香环结构的EfOM更易于和O3反应生成·OH (Sonntag et al., 2012).在模拟EfOM体系中, 研究一般以多糖、蛋白质和腐殖酸作为EfOM模拟物质开展(Lester et al., 2013).Xiong等(1992)和Ma等(1999)以腐殖酸作为EfOM模拟物研究其对臭氧氧化去除莠去津的影响, 发现当腐殖酸小于4 mg·L-1 TOC时可以显著提高氧化效果.He等(2015)以海藻酸钠、牛血清蛋白和腐殖酸模拟EfOM组成, 研究发现加入腐殖酸和牛血清蛋白后, 苯扎贝特的臭氧氧化反应受到抑制, 但反应溶液的矿化程度有所提高.
氯贝酸(Clofibric acid, CA)是一种常见的PPCPs类物质, 是调血脂药物(如安妥明、依托贝特等)的主要代谢产物.CA具有难生物降解性、高流动性, 能在环境中持久存在, 其半衰期达到21年之久(Buser et al., 1998), 在水中的滞留时间可达1~2年(Zuccato et al., 2000).目前研究对地表水、污水厂出水等样品中检测出了残留的CA, 浓度达1000 ng·L-1 (Fent et al., 2006; Gerd et al., 2010; Richardson et al., 2011).因此, 在EfOM影响PPCPs臭氧降解行为的研究中, 常以CA作为PPCPs模型污染物(Yao et al., 2016;Wang et al., 2018; Yao et al., 2018).
综上, 不同的EfOM种类及废水特性(如pH)对PPCPs的臭氧降解影响规律和机制还有待于深入研究.本文以氯贝酸(CA)为典型PPCPs, 以腐殖酸(HA)、牛血清蛋白(BSA)、海藻酸钠(AGS)和葡聚糖(Dextran)为模型EfOM, 研究不同模拟EfOM化合物对CA臭氧降解的影响及其机理, 揭示不同模拟EfOM化合物浓度和pH变化对CA臭氧降解规律的影响, 为实际二级出水中CA臭氧降解效率变化的原因分析提供理论依据、为臭氧工艺参数选择和优化提供方法指导.
2 材料与方法(Materials and methods) 2.1 试剂氯贝酸(CA)、对氯苯甲酸(p-CBA)、牛血清蛋白(BSA)、海藻酸钠(AGS)和葡聚糖(Dextran)购自美国Sigma-Aldrich公司;腐殖酸(HA)购自Acros Organics公司;所有试剂未加说明均为分析纯级.所有溶液均使用Milli-Q超纯水(18.25 MΩ·cm)配制.
2.2 实验方法 2.2.1 反应溶液的配制用超纯水配制20 mg·L-1的CA母液, 取一定量CA母液分别加入HA、BSA、AGS和Dextran溶液中, 使反应溶液中CA浓度为0.4 mg·L-1, TOC为所需浓度, 反应溶液以0.2 mol·L-1的磷酸盐缓冲液调节溶液pH, 放于冰箱冷藏备用.
2.2.2 臭氧氧化实验臭氧氧化实验在一个内径为60 mm的玻璃气泡柱反应器中以半连续方式进行.该反应器配备了恒温水浴夹层以控制反应温度恒定在25 ℃.在开始反应前, 臭氧反应器预运行30 min以达到稳定的气相O3浓度(10 mg·L-1, 101 kPa, 25 ℃), 其流速通过转子流量计控制.向反应器中加入0.4 mg·L-1的CA或p-CBA的溶液开始反应, 每组实验反应时间20 min, 取样前在样品瓶中滴入浓度为1 mol·L-1的硫代硫酸钠溶液终止臭氧反应.在反应过程中, O3气体以0.40 L·min-1的恒定流速经过反应器底部的砂芯气体分散器(平均孔径4 μm)进入500 mL反应溶液中.
2.2.3 HO·的生成量的测定HO·的生成量可通过对氯苯甲酸(p-CBA)指示物浓度的变化间接地推导得出(Elovitz et al., 1999), 其公式如下:
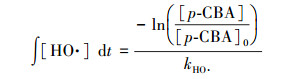
|
(1) |
式中, t为反应时间(s), [p-CBA]0为p-CBA初始浓度(0.4 mg·L-1), [p-CBA]为反应到t时间时p-CBA的浓度(mg·L-1), kHO·为p-CBA与HO·的表观反应速率常数, 取5.2×109 L·mol-1·s-1(Buxton et al., 1988).
2.3 分析方法样品在测定前均经过0.22 μm滤膜过滤, 氯贝酸和对氯苯甲酸的浓度通过高效液相色谱仪(HPLC, Dionex, Ultimate3000)测定, 色谱柱为AcclaimTM 120 C18液相色谱柱(2.2 μm, 2.1 mm ×100 mm), 流动相为1 mmol·L-1乙酸铵溶液和甲醇混合溶液(体积比为7:3), 流速为0.2 mL·min-1, 进样体积为20 μL, 检测波长为230 nm, 柱温维持在25 ℃.样品的TOC通过总有机碳分析仪(岛津TOC-L)进行测定.
3 结果与讨论(Results and discussion) 3.1 不同模拟EfOM化合物对CA臭氧降解的影响在反应溶液的初始pH值为4.0, 臭氧浓度为10 mg·L-1, 浓度折合为10 mg·L-1 TOC的不同模拟EfOM体系下, CA臭氧降解的去除效果如图 1所示.由图 1可知, 臭氧能有效降解CA, 10 min时CA去除率均超过90%.相比于超纯水对照组, HA对CA降解的促进作用较为显著, 4 min时CA去除率由80.4%提高至98.0%;而BSA抑制作用明显, 导致CA去除率从80.4%降低至60.8%.多糖类物质AGS和Dextran对CA的去除率无显著影响.
 |
| 图 1 不同模拟EfOM化合物中CA臭氧降解浓度随时间的变化 Fig. 1 CA removal during ozonation of different EfOM Model Compounds |
在不同模拟EfOM化合物中, CA的臭氧降解均符合假一级反应动力学, 如表 1所示, CA在不同EfOM中臭氧降解反应速率常数从大到小依次为HA>Dextran≈DW≈AGS>BSA, 这与去除率的规律相同.加入HA后, CA去除的反应速率常数与超纯水体系相比, 提升了3倍(从0.342 min-1提升至1.010 min-1).HA对CA臭氧降解的促进作用, 可能与HA提高HO·生成有关.
| 表 1 不同模拟EfOM化合物中CA臭氧降解的假一级反应速率常数 Table 1 Pseudo-first order rate constants for CA removal during ozonation of different EfOM Model Compounds |
-ln([p-CBA]/[p-CBA]0)与反应时间的关系如图 2所示.由该图及式(1)可计算得出HO·生成量及相关系数, 如表 2所示.AGS、HA、BSA、Dextran以及超纯水中的HO·生成量分别为1.70×10-11、2.27×10-11、0.04×10-11、0.39×10-11和2.19×10-11 mol·L-1·s.实验结果比Elovitz推测的实际二级出水中的臭氧化反应产生的HO·生成量(10-12 mol·L-1·min)高1个数量级(Elovitz et al., 2000), 可能的原因是实际二级出水中还存在着其他的HO·淬灭剂(Carbajo et al., 2015).对比超纯水和不同模拟EfOM条件下HO·的生成量可知, HA能促进HO·的生成, 而Dextran、AGS和BSA会在不同程度上抑制HO·的生成, 抑制作用规律表现为BSA>Dextran>AGS.
 |
| 图 2 不同模拟EfOM化合物中p-CBA的降解动力学 Fig. 2 Degradation kinetics of p-CBA of different EfOM Model Compounds |
| 表 2 不同模拟EfOM化合物中臭氧氧化过程HO·生成量 Table 2 HO· exposures during ozonation treatment of different EfOM Model Compounds |
HA对HO·生成的促进作用主要是由于腐殖酸类有机质中含有能与臭氧分子快速反应的活性基团(含有苯环结构的酚羟基和胺基等富电子基团), 可作为自由基引发部位, 促进臭氧分解产生HO·, 进而强化CA的臭氧降解效果.Dextran和AGS这类多糖对HO·的生成抑制作用明显, 但对CA去除效果无显著影响的现象.多糖类物质主要是脂肪族饱和结构, 在臭氧氧化体系中, 其与臭氧分子的反应速率较慢(kO3=0.45 L·mol-1·s-1(Hoigne et al., 1983)), 但会消耗反应过程中的HO·, 生成反应活性更强的O2·-, 如式(2)~(4)所示(Akhlaq et al., 1990), 并可通过O2·-氧化降解CA.因此, 与超纯水相比, Dextran和AGS类多糖会造成HO·生成量的减少, 但对CA臭氧降解的抑制作用不明显.
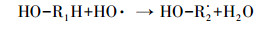
|
(2) |
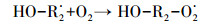
|
(3) |

|
(4) |
已有研究发现, EfOM会影响PPCPs的臭氧降解, 但作用效果取决于EfOM的含量(Huber et al., 2005; Yao et al., 2016).因此, 进一步探究了不同HA及BSA浓度对CA臭氧降解和HO·生成的影响情况.
3.3.1 腐殖酸的影响反应溶液的初始pH值为4.0, 臭氧投加量为5 mg·L-1时, 不同HA折合TOC浓度对HO·生成和CA降解的影响如表 3、图 3所示, 随着HA浓度的增加, HO·生成量和CA降解效果趋势一致.在1 mg·L-1 TOC的低浓度条件下, HA对CA降解促进作用最为显著.与无HA添加时相比, CA的去除率相近, 二者均能达到100%, 但反应速率常数提高了1.95倍(从0.588 min-1提高到1.148 min-1).但当HA浓度提高到10 mg·L-1 TOC时, 与无HA添加时相比, 去除率降低了10.6%, 反应速率常数也从0.588 min-1降低至0.376 min-1.这一现象表明, HA的存在对CA臭氧降解的影响比较复杂, 一方面, 较低浓度的腐殖酸可以作为HO·链式反应的引发剂、促进剂, 加速臭氧分解产生HO·;另一方面, 过高浓度的腐殖酸也可成为HO·的捕获剂或与CA竞争臭氧分子(芮旻等, 2006).
| 表 3 不同浓度HA下臭氧降解过程中HO·的生成 Table 3 HO· exposures during ozonation in the presence of different concentrations of HA |
 |
| 图 3 不同浓度HA对CA臭氧降解效果的影响 Fig. 3 Ozonation of CA removal in the presence of different concentrations of HA |
| 表 4 不同浓度HA下CA臭氧降解的假一级反应速率常数 Table 4 Pseudo-first order rate constants for CA removal during ozonation of different concentrations of HA |
亦有文献报道, 莠去津臭氧降解过程中, 当腐殖酸浓度小于4 mg·L-1 TOC时可以显著提高降解水平, 降解效果最佳的HA浓度为1 mg·L-1 TOC(Xiong et al., 1992), 与本实验结果相类似.
3.3.2 BSA的影响BSA存在对HO·生成量和CA臭氧降解有抑制作用, 并且抑制作用随BSA浓度的增加而增加, 当BSA浓度从0增加到10 mg·L-1 TOC时, CA的去除率从100.0%降低至48.9%, 臭氧降解反应速率常数从0.588 min-1降低至0.064 min-1.BSA在结构上由不同的氨基酸组成, 有文献研究表明, 特定的氨基酸与臭氧分子和HO·都有较高的反应速率(Marc-Olivier et al., 2006), 因此BSA会与CA竞争臭氧分子和HO·, 进而抑制CA的臭氧降解效果.
| 表 5 不同浓度BSA下臭氧降解过程中HO·的生成 Table 5 HO· exposures during ozonation treatment in the presence of different concentrations of BSA |
水体pH值的变化会改变水体中有机物的存在形式, 也会影响O3的分解和HO·的生成, 进而对污染物的降解产生重要影响(Xiong et al., 2018).为研究pH对CA臭氧降解的影响, 配置了不同初始pH值(4、7、10)的AGS、HA、BSA和Dextran溶液进行CA臭氧降解实验, 结果如图 5所示.实验结果表明, 超纯水中, HO·生成量和CA臭氧降解效率均随着pH的增加而增大.CA的pKa为3.2(Rosal et al., 2010), 本实验条件下的pH值不会影响其解离状态.因此, pH值增长可对臭氧氧化效果产生强化作用, 主要是因为OH-可促进O3分解(Andreozzi et al., 2003).在4种模拟EfOM化合物中, HO·生成量和CA臭氧降解效率也随着pH的增加而增大.
 |
| 图 4 不同浓度BSA对CA臭氧降解效果的影响 Fig. 4 Ozonation of CA removal in the presence of different concentrations of BSA |
| 表 6 不同浓度BSA下CA臭氧降解的假一级反应速率常数 Table 6 Pseudo-first order rate constants for CA removal during ozonation of different concentrations of BSA |
 |
| 图 5 pH值对不同模拟EfOM化合物中CA臭氧降解过程中HO·生成量的影响(a)和CA臭氧降解的影响(b) Fig. 5 The effect of pH on the HO· exposures(a) and the ozonation of CA(b) in the presence of different EfOM |
研究发现溶液pH会影响HA对CA臭氧降解的作用效果.pH=4时, HA投加可以促进HO·生成和臭氧降解, 但随着pH的升高, HA则对其表现出抑制作用.这主要是因为在酸性条件下, HA以分子的形式存在, 其结构中除含有大量的苯环外, 还含有羧基、羰基和酚羟基等官能团(Westerhoff et al., 1999), 易于和臭氧反应生成HO·, 从而促进CA的臭氧降解反应, 而在高pH条件下, 腐殖酸分子呈线性结构, 荧光强度增强, 芳香性更大, 导致其对HO·的捕获作用大于促进其生成作用(Au et al., 1999, 傅平青等, 2004).因此在较高pH条件下, HA会对CA臭氧降解效果产生抑制作用.
4 结论(Conclusions)1) 不同模拟EfOM化合物中, CA臭氧降解符合假一级反应动力学, 臭氧降解效果呈现出HA>Dextran≈DW≈AGS>BSA的规律, HA可提高HO·的生成速率从而加快CA降解效率, 使反应速率常数提升3倍, 去除率提高17.5%;而BSA则明显抑制HO·的生成从而降低CA降解速率.
2) HA对CA臭氧降解的影响具有双重性:HA浓度较低时, 其可作为HO·链式反应的引发剂、促进剂, 强化CA去除, 例如当HA浓度为1 mg·L-1TOC时, 与DW体系相比, CA去除的反应速率常数从0.588 min-1提高到1.148 min-1;而HA浓度较高时, 其会捕获HO·并与CA竞争臭氧分子, 降低处理效果, 例如当HA浓度为10 mg·L-1 TOC时, 与DW体系相比, 反应速率常数降低至0.376 min-1.
3) 不同模拟EfOM化合物投加时HO·生成量和CA臭氧降解效率均随着pH的增加而增大.溶液pH值会影响HA对CA臭氧降解的作用效果:pH=4时, HA投加可以促进CA降解和HO·生成;但pH为7或9时, 因HA的存在形态发生了改变, 其对CA臭氧降解效果表现出抑制作用.
Akhlaq M S, Schuchmann H P, Sonntag C V. 1990. Degradation of the polysaccharide alginic acid: A comparison of the effects of UV light and ozone[J]. Environmental Science & Technology, 24(3): 379–383.
|
Andreozzi R, Caprio V, Marotta R, et al. 2003. Ozonation and H2O2/UV treatment of clofibric acid in water: A kinetic investigation[J]. Journal of Hazardous Materials, 103(3): 233–246.
DOI:10.1016/j.jhazmat.2003.07.001
|
Au K K, Penisson A C, Yang S, et al. 1999. Natural organic matter at oxide/water interfaces: Complexation and conformation[J]. Geochimica Et Cosmochimica Acta, 63(19/20): 2903–2917.
|
Audenaert W T, Vandierendonck D, Van S H, et al. 2013. Comparison of ozone and HO· induced conversion of effluent organic matter (EfOM) using ozonation and UV/H2O2 treatment[J]. Water Research, 47(7): 2387–2398.
DOI:10.1016/j.watres.2013.02.003
|
Buser H R, Müller M D, Theobald N. 1998. Occurrence of the pharmaceutical drug clofibric acid and the herbicide mecoprop in various Swiss Lakes and in the North Sea[J]. Environmental Science & Technology, 32(1): 188–192.
|
Buxton G V, Greenstock C L, Helman W P, et al. 1988. Critical review of rate constants for reactions of hydrated electrons, hydrogen-atoms and hydroxyl radicals (.OH/.O-) in aqueous-solution[J]. Journal of Physical and Chemical Reference Data, 17(2): 513–886.
DOI:10.1063/1.555805
|
Carbajo J B, Petre A L, Rosal R, et al. 2015. Continuous zonation treatment of ofloxacin: Transformation products, water matrix effect and aquatic toxicity[J]. Journal of Hazardous Materials, 292: 34–43.
DOI:10.1016/j.jhazmat.2015.02.075
|
Elovitz M S, von Gunten U. 1999. Hydroxyl radical ozone ratios during ozonation processes. Ⅰ-The R-ct concept[J]. Ozone-Science & Engineering, 21: 239–260.
|
Elovitz M S, von Gunten U, Kaiser H P. 2000. Hydroxyl radical/ozone ratios during ozonation processes. Ⅱ. The effect of temperature, pH, alkalinity, and DOM properties[J]. Ozone-Science & Engineering, 22(2): 123–150.
|
Fent K, Weston A A, Caminada D. 2006. Ecotoxicology of human pharmaceuticals[J]. Aquatic Toxicology, 76(2): 122–159.
|
傅平青, 刘丛强, 尹祚莹, 等. 2004. 腐殖酸三维荧光光谱特性研究[J]. 地球化学, 2004, 33: 301–308.
|
Gerd H, Hansen P D, Maurer H H, et al. 2010. Environmental risk assessment of medicinal products for human use according to European Commission recommendations[J]. Environmental Toxicology, 19(3): 226–240.
|
He H, Sui Q, Lu S, et al. 2015. Effect of effluent organic matter on ozonation of bezafibrate[J]. Frontiers of Environmental Science & Engineering, 9(6): 962–969.
|
Hoigne J, Bader H. 1983. Rate constants of reactions of ozone with organic and inorganic compounds in water—Ⅰ: Non-dissociating organic compounds[J]. Water Research, 17(2): 173–183.
|
Huber M M, Gobel A, Joss A, et al. 2005. Oxidation of pharmaceuticals during ozonation of municipal wastewater effluents: A pilot study[J]. Environmental Science & Technology, 39(11): 4290–4299.
|
Jun M A, Graham N J D. 1999. Degradation of atrazine by manganese-catalysed ozonation: Influence of humic substances[J]. Water Research, 33(3): 785–793.
DOI:10.1016/S0043-1354(98)00266-8
|
Lester Y, Avisar D, Mamane H. 2013. Ozone degradation of cyclophosphamide effect of alkalinity and key effluent organic matter constituents[J]. Ozone-Science & Engineering, 35(2): 125–133.
|
Marc-Olivier B, Urs V G. 2006. Phenols and amine induced HO· generation during the initial phase of natural water ozonation[J]. Environmental Science & Technology, 40(9): 3057–3063.
|
Michael I, Rizzo L, McArdell C S, et al. 2013. Urban wastewater treatment plants as hotspots for the release of antibiotics in the environment: A review[J]. Water Research, 47(3): 957–995.
DOI:10.1016/j.watres.2012.11.027
|
Nakada N, Shinohara H, Murata A, et al. 2007. Removal of selected pharmaceuticals and personal care products (PPCPs) and endocrine-disrupting chemicals (EDCs) during sand filtration and ozonation at a municipal sewage treatment plant[J]. Water Research, 41(19): 4373–4382.
DOI:10.1016/j.watres.2007.06.038
|
Nikolaou A, Meric S, Fatta D. 2007. Occurrence patterns of pharmaceuticals in water and wastewater environments[J]. Analytical & Bioanalytical Chemistry, 387(4): 1225–1234.
|
Ribeiro A R, Nunes O C, Pereira M F R, et al. 2015. An overview on the advanced oxidation processes applied for the treatment of water pollutants defined in the recently launched Directive 2013/39/EU[J]. Environment International, 75: 33–51.
DOI:10.1016/j.envint.2014.10.027
|
Richardson M L, Bowron J M. 2011. The fate of pharmaceutical chemicals in the aquatic environment[J]. Journal of Pharmacy & Pharmacology, 37(1): 1–12.
|
Rivera-Utrilla J, Sanchez-Polo M, Ferro-Garcia M A, et al. 2013. Pharmaceuticals as emerging contaminants and their removal from water. A review[J]. Chemosphere, 93(7): 1268–1287.
DOI:10.1016/j.chemosphere.2013.07.059
|
芮旻, 高乃云, 徐斌, 等. 2006. 水中腐殖酸对高级氧化联用技术去除内分泌干扰物(DMP)的影响[J]. 环境科学, 2006, 27(12): 2495–2501.
DOI:10.3321/j.issn:0250-3301.2006.12.023 |
Rosal R, Rodriguez A, Antonio Perdigon-Melon J, et al. 2010. Occurrence of emerging pollutants in urban wastewater and their removal through biological treatment followed by ozonation[J]. Water Research, 44(2): 578–588.
DOI:10.1016/j.watres.2009.07.004
|
Sonntag C V, Gunten U V. 2012. Chemistry of ozone in water and wastewater treatment[M]. London: IWA publishing
|
宋存义, 汪翠萍, 李晖. 2009. 污水处理中几种去除药物及个人护理用品方法的机理及效果比较[J]. 环境工程学报, 2009, 3(11): 1921–1930.
|
Wang H, Zhan J, Yao W, et al. 2018. Comparison of pharmaceutical abatement in various water matrices by conventional ozonation, peroxone (O3/H2O2), and an electro-peroxone process[J]. Water Research, 130: 127–138.
DOI:10.1016/j.watres.2017.11.054
|
Westerhoff, Aiken G, Amy G, et al. 1999. Relationships between the structure of natural organic matter and its reactivity towards molecular ozone and hydroxyl radicals[J]. Water Research, 33(10): 2265–2276.
DOI:10.1016/S0043-1354(98)00447-3
|
Xiong F, Graham N J D. 1992. Removal of atrazine through ozonation in the presence of humic substances[J]. Ozone-Science & Engineering, 14: 263–268.
|
Xiong Z, Lai B, Yang P. 2018. Insight into a highly efficient electrolysis-ozone process for N, N -dimethylacetamide degradation: Quantitative analysis of the role of catalytic ozonation, Fenton-like and peroxone reactions[J]. Water Research, 140: 12–23.
DOI:10.1016/j.watres.2018.04.030
|
Yao W, Rehman S W U, Wang H, et al. 2018. Pilot-scale evaluation of micropollutant abatements by conventional ozonation, UV/O3, and an electro-peroxone process[J]. Water Research, 138: 106–117.
DOI:10.1016/j.watres.2018.03.044
|
Yao W, Wang X, Yang H, et al. 2016. Removal of pharmaceuticals from secondary effluents by an electro-peroxone process[J]. Water Research, 88: 826–835.
DOI:10.1016/j.watres.2015.11.024
|
Zuccato E, Calamari D, Natangelo M, et al. 2000. Presence of therapeutic drugs in the environment[J]. Lancet, 355(9217): 1789–1790.
DOI:10.1016/S0140-6736(00)02270-4
|
周海东, 黄霞, 文湘华. 2007. 城市污水中有关新型微污染物PPCPs归趋研究的进展[J]. 环境工程学报, 2007, 1(12): 1–9.
DOI:10.3969/j.issn.1673-9108.2007.12.001 |