 2019, Vol. 39
2019, Vol. 39



近年来, 基于硫酸根自由基(SO4-·)的高级氧化技术在环境污染治理领域受到广泛关注(Gao et al., 2012;Richard et al., 2015).SO4-·不仅具有较高的氧化还原电位(E0=2.5 ~3.1 V), 而且在水溶液中的半衰期较长, 能氧化降解大部分有机污染物(郑垒等, 2016).SO4-·主要通过热活化(Gao et al., 2016)、光活化(Wang et al., 2015)、过渡金属活化(Wu et al., 2017)以及电活化(Liu et al., 2018)等方式破坏过硫酸盐(PDS)的氧氧键(—O—O—键)产生.然而现有的活化方式均存在一定的缺陷.以电活化为例, 电活化利用阴极电极发生的还原反应, 使PDS得电子生成SO4-·.反应过程需要额外的电能供应, 易造成能耗问题.
微生物燃料电池(MFC)是一种绿色发电技术(曹效鑫等, 2006).阳极厌氧微生物在降解底物后会产生电子, 电子经外电路传递到阴极, 阴极电子受体接受电子, 该过程中电子不断被产生、传递、接受, 从而形成电流(尤世界等, 2006).将PDS作为电子受体置于阴极室, PDS接受电子后发生电活化反应生成SO4-·, 实现MFC活化PDS.该活化过程能避免传统电活化的能耗问题;而且由于PDS在水溶液中溶解度高、传质系数大、标准电极电势高等特点能有效提高MFC产电性能(付乾等, 2009).
铜作为常见的过渡金属在催化反应领域得到广泛研究.张剑桥(2016)研究发现Cu2+能有效强化Fe2+活化PDS降解苯酚, 推测反应机理是Cu2+被还原成Cu+, Cu+能活化PDS, 促进SO4-·生成, 进而提高污染物降解率.铜离子除了强化PDS活化体系对污染物的降解之外, 还能提高MFC产电性能.牟姝君等(2014)研究铜离子对双室MFC的产电影响.结果表明, 阴极500 mg·L-1的铜离子可显著降低阴极反应的活化内阻和总体表观内阻, 提高MFC产电性能.然而, 目前尚未有关于CuO同时实现强化PDS活化体系及MFC产电性能的研究报道.
本研究以甲基橙(MO)为模拟污染物, 探索CuO强化MFC活化PDS(CuO/MFC/PDS体系)及同步产电性能的可行性.考察初始pH、PDS浓度、CuO浓度等因素对CuO/MFC/PDS体系降解MO及同步产电性能的影响, 并对其降解机理和矿化程度进行分析.
2 材料与方法(Materials and methods) 2.1 实验装置实验装置是由有机玻璃制成的传统“H型”双室MFC.阳极室和阴极室等体积(5 cm×5 cm×8 cm), 两室之间以质子交换膜(Nafion-117, 杜邦公司)隔绝开.阳极和阴极的电极材料均为碳毡(长宽均为3 cm, 厚度0.3 cm, 北京碳电厂), 阳极电极和阴极电极通过钛丝经外电路串联外载电阻形成回路.装置搭建前质子交换膜需进行如下预处理以去除其表面的有机杂质:在80 ℃下, 3% H2O2中加热1 h, 然后依次于80 ℃蒸馏水、5 mol·L-1 H2SO4中各浸泡1 h, 用蒸馏水反复冲洗至冲洗液呈中性为止, 最后置于蒸馏水中备用(孔晓英等, 2016).
 |
| 图 1 装置示意图 Fig. 1 Schematic diagram of the device |
红螺菌作为一种兼性营养型细菌, 普遍存在于池塘、湖泊淤泥中, 目前已被应用于降解高浓度有机废水.本研究选用深红红螺菌397(台湾帝翰生物科技有限公司)作为阳极产电微生物.阳极液:1 g·L-1 NH4Cl, 1 g·L-1 NaHCO3, 0.2 g·L-1 K2HPO4, 2 g·L-1 CH3COONa, 0.2 g·L-1 MgSO4·7H2O, 1 g·L-1 NaCl, 10 mL·L-1微量元素溶液, pH为7.0.阴极液:2 mmol·L-1 PDS溶液, pH为3.0.阳极液和阴极液体积均为120 mL, 连通电路后开始进行阳极产电微生物的挂膜驯化, 在运行过程中阳极室始终保持厌氧环境.将20通数据采集器(34970A, 美国安捷伦科技公司)连接于外载电阻两端, 测量MFC输出电压.当输出电压小于50 mV时, 需更换阳极液和阴极液(骆海萍等, 2008), 视为完成1个周期.连续运行3~4个周期后, 完成阳极产电微生物的挂膜驯化, MFC启动成功, 此时输出电压基本达到稳定状态.
最后在保持阳极液不变的条件下, 将阴极液更换为MO溶液, 并投加PDS及CuO.MO浓度设定为0.1 mmol·L-1, PDS浓度和CuO浓度根据不同因素实验中的反应量决定.
2.3 分析测试方法 2.3.1 水质指标采用分光光度法测定MO降解率.由于MO在不同pH下有偶氮和蒽醌两种结构式, 其紫外-可见吸收光谱的最大吸收波长会随pH变化而变化(Liu et al., 2009;李明玉等, 2009), 但当pH≥5.0后最大吸收波长固定在468 nm.为了准确测定MO降解率, 每次取1 mL MO水样后加入1 mL甲醇用于淬灭自由基(毕晨等, 2017), 然后用碱液稀释至pH≥5.0(李霞, 2017), 最后经0.45 μm滤膜过滤后, 在468 nm处测定吸光度.根据实验所得吸光度与浓度之间的标准曲线(y=0.0743x+0.0295), 计算MO浓度及降解率.
TOC采用总有机碳分析仪(Multi N/C2100, 德国耶拿)测定, 样品需先经0.45 μm滤膜过滤.实验以氧气作为载气, 气体流量设置为(160±10) mL·min-1, 温度设置为800 ℃.
紫外-可见吸收光谱(UV-vis)采用紫外可见分光光度计(specord50, 德国耶拿)进行扫描测定.扫描波长范围为200~700 nm, 狭缝宽度为1 nm, 扫描速度为50 nm·s-1.
2.3.2 产电指标电流密度按式(1)计算:
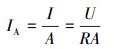
|
(1) |
式中, IA为电流密度(mA·m-2);I为电流(mA);U为外载电阻两端的电压值, 由数据采集器直接记录保存(mV);R为外载电阻的电阻值, 在无特殊说明的情况下, 其电阻值均为1000 Ω;A为阳极电极的有效表面积(m2).
功率密度按式(2)计算:
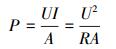
|
(2) |
式中, P为功率密度(mW·m-2).
表观内阻采用稳定放电法测定(梁鹏等, 2007).通过测量不同外载电阻(30000~100 Ω)条件下, MFC稳定放电时各电阻对应的输出电压, 以电流为横坐标, 电压为纵坐标绘制极化曲线.通过对极化曲线进行线性拟合, 所得斜率的绝对值即为表观内阻.
3 结果与讨论(Results and discussion) 3.1 MFC启动阶段MFC启动阶段电压变化如图 2所示.启动初期, 随着微生物降解底物后活性提高和阳极电极上微生物的富集, MFC电压持续增长, 220 min时达到最高376.7 mV.由于阳极底物和阴极PDS被消耗, 此后电压逐步下降至50 mV.更换阳极液和阴极液后连续运行4周期, MFC启动完成, 电压基本稳定在377 mV.
 |
| 图 2 MFC启动阶段电压变化(箭头表示添加PDS) Fig. 2 Voltage change during the MFC startup (arrow indicating the replacement of catholyte) |
实验考察不同反应体系下MO降解率.由图 3可知, MFC阴极可降解MO, 反应4 h降解率达59.6%.Liu等(2009)研究发现, MO可作电子受体在阴极接受电子, 发生四电子还原反应(式(3)), 实现降解.
 |
| 图 3 不同体系下MO降解率 Fig. 3 MO degradation rate under different conditions |
MFC/PDS体系内MO降解率为86.5%.与MFC阴极降解MO不同的是, MO降解并非还原反应所致, 而是被SO4-·氧化降解.付乾等(2009)研究表明, PDS作阴极电子受体, 会发生单电子还原反应生成SO4-·, 其氧化还原电位较高, 能有效氧化降解MO.
CuO/MFC/PDS体系内MO降解率最佳, 为99.3%.与MFC+PDS体系相比, 上升幅度达12.8%.由图 3可知, 单独投加CuO时MO降解率仅1.8%, 可判断MO降解率升高并非CuO吸附所致.推测是由于CuO在MFC阴极得电子会生成Cu+, Cu+能活化PDS生成SO4-·(式(4)和(5)), 提高SO4-·稳态浓度, 促进MO降解.可见CuO对MFC/PDS体系存在促进作用.
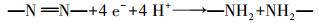
|
(3) |
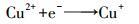
|
(4) |
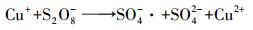
|
(5) |
在溶液初始pH为3.0, PDS浓度为2 mmol·L-1时, 测定不同CuO浓度(0、0.2、0.4、0.6 mmol·L-1)下MO降解率.如图 4所示, CuO浓度从0 mmol·L-1增加到0.2 mmol·L-1时, MO降解率从86.5%升高到93.3%.当浓度进一步增加到0.6 mmol·L-1时, 降解率达到最高99.3%, 比未投加CuO时升高12.8%.
 |
| 图 4 CuO浓度对MO降解率的影响 Fig. 4 Effect of CuO concentration on the degradation rate of MO |
推测是因为CuO在MFC阴极得电子会生成Cu+, Cu+能活化PDS生成SO4-·(式(3)和(4)).增加CuO浓度可以提高Cu+数量, 从而促进SO4-·生成, MO降解率也随之升高.唐琪等(2017)研究发现, CuO/PDS体系中CuO浓度过高会抑制被活化的PDS与污染物接触反应, 影响污染物降解.但是本研究未观察到类似现象, 推测是因为CuO最高浓度(0.6 mmol·L-1)尚未过量.
3.3.2 CuO浓度对产电性能的影响有研究报道(牟姝君等, 2014), 双室MFC阴极室添加铜离子可以显著降低体系内阻, 提高产电效率.但当阴极铜离子浓度过高时, 部分铜离子会迁移到阳极室, 影响产电微生物活性及体系产电性能.本研究通过测定各CuO浓度下MFC输出功率密度及表观内阻来判断其浓度对产电性能的影响.
在3.3.1节相同条件下测定体系的产电性能.如图 5a所示, 当CuO浓度从0 mmol·L-1增加到0.6 mmol·L-1时, MFC最大输出功率密度从53.0 mW·m-2增大到103.5 mW·m-2;由图 5b可知, 体系对应的表观内阻从484.1 Ω减小到318.6 Ω.推测理由如下:①增加CuO浓度, 可以促进阴极室自由基的生成, 其较高的氧化还原电位和活性, 有利于提高体系产电性能(付乾等, 2009);②CuO附着在阴极电极上, 其表面的Cu2+能提高阴极电极电势, 增加溶液导电率, 扩大阴阳极的电势差, 提高产电性能(朱美月等, 2017).
 |
| 图 5 CuO浓度对功率密度(a)和表观内阻(b)的影响 Fig. 5 Effect of CuO concentration on power density (a) and apparent internal resistance (b) |
以上研究结果与其他研究文献(Miran et al., 2017;朱美月等, 2017)结果基本一致.但不同的是, 过高的铜离子浓度并未出现影响阳极产电微生物活性, 抑制体系产电性能的现象, 可能是因为阴极室反应主要是在阴极电极和CuO表面发生, 不存在过多Cu2+溶于水体, 阴阳极的铜离子浓度梯度较低, 阴极Cu2+未发生迁移.
综上, MO降解率及同步产电性能随CuO浓度变化表现出相同的变化趋势, 两者均随浓度增大而增大.推测可能存在如下关系:增加CuO浓度, 能促进Cu+的生成, 提高SO4-·稳态浓度, 增强同步产电性能, 产电性能的提高也意味着阳极传递到阴极的电子数量增多.MFC阴极电子受体不唯一, 除Cu2+之外, PDS也能作为电子受体接受电子, 发生单电子还原反应生成SO4-·(式(6)).当电子传递数量增多时, SO4-·增多, 体系氧化性增强, MO降解率提高.
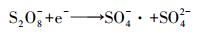
|
(6) |
在CuO浓度为0.6 mmol·L-1, 初始pH为3.0时, 测定不同PDS浓度(1、2、3、4 mmol·L-1)下MO降解率.如图 6所示, PDS浓度从1 mmol·L-1增加到2 mmol·L-1时, MO降解率从88.4%增大到99.3%.但PDS浓度进一步增加到3 mmol·L-1时, MO降解率却开始受到抑制.这是因为增加PDS浓度, 有利于SO4-·生成(式(5)和(6)), 提高MO降解率(Zhang et al., 2014).但PDS浓度过高(大于2 mmol·L-1)时, 多余的PDS会与SO4-·发生淬灭反应(式(7)), 减少自由基数量(Nie et al., 2015), 降低体系整体氧化能力, 从而导致MO降解率下降.
 |
| 图 6 PDS浓度对MO降解率的影响 Fig. 6 Effect of PDS concentration on the degradation rate of MO |

|
(7) |
在3.4.1节相同条件下测定体系的产电性能.如图 7a和7b所示, PDS浓度从1 mmol·L-1增加到2 mmol·L-1时, 体系最大输出功率密度从37.0 mW·m-2增大到103.5 mW·m-2, 对应体系的表观内阻从647.1 Ω减小到318.6 Ω.当PDS浓度增加到4 mmol·L-1时, 最大输出功率密度上升至114.7 mW·m-2, 上升幅度较小;与CuO浓度为2 mmol·L-1时相比, 表观内阻却增大到338.6 Ω.说明PDS浓度的增加有利于提高体系产电性能, 但是PDS浓度过高时, 会出现抑制作用.
 |
| 图 7 PDS浓度对功率密度(a)和表观内阻(b)的影响 Fig. 7 Effect of PDS concentration on power density (a) and apparent internal resistance (b) |
初步推测PDS作为阴极室的电子受体, 其浓度的增加一方面会提高阴极电极电势(由能斯特方程可知);另一方面会扩大阴极电极与阴极室内PDS的浓度差, 增大PDS向阴极电极上扩散的驱动势, 加剧阴极反应, 提高产电性能(付乾等, 2009).但PDS浓度过高时, 多余的PDS会与阴极室内自由基发生淬灭反应, 影响PDS得电子, 降低阴阳极之间的电子传递效率, 所以体系产电性能开始下降.
综上, MO降解率及产电性能均随PDS浓度的增加先增大后减小.推测MFC产电性能对MO降解率具有一定影响.当产电提高时, 阳极传递到阴极的电子数量增多, 阴极Cu+生成较多, 导致SO4-·生成数增多, MO降解率升高.
3.5 初始pH的影响 3.5.1 初始pH对MO降解率的影响初始pH是过硫酸盐活化体系的重要影响因素.本研究在CuO浓度为0.6 mmol·L-1, PDS浓度为2 mmol·L-1时, 测定不同初始pH(2.0、3.0、4.0、5.0)下MO降解率.如图 8所示, 初始pH从5.0降低到3.0时, MO降解率从78.6%增大到99.3%.当初始pH降到2.0时, 降解率略微降低至98.4%.可见初始pH的降低有利于MO降解, 但并非越低越好.
 |
| 图 8 初始pH对MO降解率的影响 Fig. 8 Effect of initial pH on the degradation rate of MO |
一方面是由于初始pH的降低会减少溶液中溶解氧含量(Chen et al., 2015), 缓解氧气与PDS得电子形成的竞争, 促进PDS得电子产生SO4-·;另一方面酸性条件利于CuO中Cu2+的释放, 释放的Cu2+被MFC阴极还原为Cu+, Cu+与PDS反应生成SO4-·.所以初始pH的降低会促进MO降解.但初始pH过低时, 会发生酸催化反应(高乃云等, 2013), 体系内SO4-·生成过多(式(8)和(9)), 互相发生淬灭反应(式(10)), 抑制MO降解.
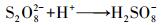
|
(8) |
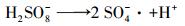
|
(9) |
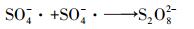
|
(10) |
在3.5.1节相同条件下测定体系的产电性能.如图 9所示, 各初始pH下体系最大输出功率密度分别为126.2、103.5、65.0和39.3 mW·m-2, 对应的表观内阻分别为586.4、453.0、318.6和283.4 Ω.初始pH的降低会增大体系的最大输出功率密度, 减小表观内阻.一方面可能是因为初始pH的降低会提高阴极电极电势(Li et al., 2009);另一方面初始pH的降低会提高阴极H+浓度, 体系内发生酸催化反应促进自由基的生成.考虑到自由基具有较高的氧化还原电位和活性, 体系产电性能才会得到提高.
 |
| 图 9 初始pH对功率密度(a)和表观内阻(b)的影响 Fig. 9 Effect of initial pH on power density (a) and apparent internal resistance (b) |
综上, 初始pH在3.0~5.0之间时, MO降解率与MFC产电性能均随初始pH的降低而增大.初步推测是因为初始pH降低会增强MFC产电性能, 提高MFC阳极传递到阴极参与活化反应的电子数量, 增加SO4-·浓度, 促进MO降解.而当初始pH从3.0降至2.0时, MFC产电性能随之升高, MO降解率却呈下降趋势.可能是由于MFC产电性能升高后, 电子传递过多, SO4-·数量激增, 相互之间发生淬灭反应, 从而抑制MO降解.
3.6 MO降解的反应动力学为探索CuO强化MFC活化PDS体系降解MO的反应动力学.本研究对不同反应条件下的降解率进行动力学拟合.
| 表 1 不同反应条件下的反应速率常数 Table 1 Reaction rate constants under different reaction conditions |
拟合结果表明, 体系对MO的降解符合准一级动力学模型.各条件下反应速率常数如下表所示.当CuO浓度从0 mmol·L-1增加到0.6 mmol·L-1, 反应速率常数从0.008 min-1增大到0.019 min-1.CuO浓度的增加有利于Cu+的生成, 提高SO4-·稳态浓度, 促进MO降解.当PDS浓度从1 mmol·L-1到4 mmol·L-1, 对应反应速率常数分别为0.009、0.019、0.012和0.016 min-1.PDS浓度的升高, 有利于SO4-·生成, 但PDS浓度过高时, SO4-·会相互淬灭, 抑制MO降解.当初始pH为2.0到5.0时, 反应速率常数分别为0.016、0.019、0.009和0.006 min-1.初始pH越低, 反应速率常数越高.初始pH降低, H+浓度升高, 有利于SO4-·生成, 促进MO降解.
3.7 降解机理研究诸多研究表明(Ghauch et al., 2012;Lin et al., 2013), 过硫酸盐活化体系中的主要活性物质为SO4-·和·OH.本研究在最佳反应条件下分别投加20 mmol·L-1甲醇和叔丁醇, 通过比较MO降解受到的抑制程度来判断体系内起主导作用的活性物质.甲醇由于含有α—H键, 其对SO4-·和·OH均有较好的淬灭效果;叔丁醇不含有α—H键, 其与·OH的反应速率常数是与SO4-·反应速率常数的1000倍, 只能淬灭·OH.
如图 10所示, 投加甲醇和叔丁醇后MO降解率分别为55.1%和85.1%, 下降了44.2%和14.2%.两种淬灭剂均能抑制MO降解, 且甲醇的抑制程度较叔丁醇高出很多.因此, 可以判断体系内的主要活性物质为SO4-·和少量·OH.
 |
| 图 10 淬灭剂对MO降解率的影响 Fig. 10 Effect of quencher on the degradation rate of MO |
为研究MO降解机理, 对投加CuO前后的MO降解过程进行紫外-可见光扫描.结果如图 11所示, 投加CuO前后, MO降解过程中468 nm处特征吸收峰均随着反应时间的推移不断减小, 且投加CuO后MO降解过程中的特征吸收峰下降幅度更大.该处吸收峰是由MO发色基团偶氮键(—N=N—键)n-π*跃迁引起的, 吸收峰降低对应于MO降解率, 因此可以判断投加CuO有利于MO发色基团偶氮键的断裂, 促进MO降解.
 |
| 图 11 投加CuO前(a)、后(b)MO降解过程的紫外-可见光谱 Fig. 11 UV-visible spectrum of MO degradation process |
同时由图 11b可知, 从0 min到180 min时, 275 nm处吸收峰不断增长, 该处吸收峰是由苯环共轭体系的π-π*跃迁引起的.推测是MO降解过程中产生含有苯环类的中间产物.当反应到240 min时, 吸收峰开始下降, 表明苯环结构开环, 中间产物被降解.
孔令国等(2010)研究表明, MO结构中偶氮键的断裂和苯环结构的开环分别影响着MO降解率和矿化率.结合图 11、12可知, MO降解过程中偶氮键容易断裂, 降解速率快;但苯环结构由于化学性质相对稳定, 难以开环, MO矿化速率低.为判断体系对MO矿化率, 在最佳反应条件下, 测定MO总有机碳含量(TOC).如图 13所示, 60 min时, TOC去除率达到18.1%, 而180 min, TOC去除率达27.5%, 上升幅度仅为9.4%, 这是由于难降解中间产物的生成所致.反应240 min后, TOC去除率达到36.6%.表明体系对MO具有一定的矿化能力.
 |
| 图 12 MO可能的降解途径 Fig. 12 Possible degradation pathways for MO |
 |
| 图 13 MO降解过程的TOC去除率 Fig. 13 TOC removal rate in the process of MO degradation |
1) CuO能有效提高MFC活化PDS体系中MO降解率.最佳反应条件:初始pH为3.0, CuO浓度为0.6 mmol·L-1, PDS浓度为2 mmol·L-1时, 反应4 h后MO降解率达到99.3%, 比未投加CuO时MO降解率提高12.8%.动力学研究表明, 降解过程符合准一级动力学模型.
2) CuO的投加对体系同步产电性能有较大促进作用.MFC最大输出功率密度从53.0 mW·m-2增大到103.5 mW·m-2, 输出功率密度提高1.95倍;表观内阻从484.1 Ω减小到318.6 Ω, 下降幅度达到34.1%.
3) 自由基淬灭实验表明, MO降解过程中的主要活性物质为SO4-·和少量·OH.
4) 紫外-可见光谱对比显示, MO降解过程中偶氮键率先断裂, 然后生成含苯环类的中间产物, 最终得到矿化, 矿化率为36.6%.
毕晨, 施周, 周石庆, 等. 2017. EGCG强化Fe2+/过硫酸盐体系降解金橙G的研究[J]. 中国环境科学, 2017, 37(10): 3722–3728.
DOI:10.3969/j.issn.1000-6923.2017.10.013 |
Chen W S, Huang C P, Chin P, et al. 2015. Mineralization of aniline in aqueous solution by electrochemical activation of persulfate[J]. Chemosphere, 125: 175–181.
DOI:10.1016/j.chemosphere.2014.12.053
|
曹效鑫, 梁鹏, 黄霞, 等. 2006. "三合一"微生物燃料电池的产电特性研究[J]. 环境科学学报, 2006, 26(8): 1252–1257.
DOI:10.3321/j.issn:0253-2468.2006.08.005 |
Cao Y Q, Gao N Y, Deng Y, et al. 2012. Ultraviolet (UV) light-activated persulfate oxidation of sulfamethazine in water[J]. Chemical Engineering Journal, 195: 248–253.
|
付乾, 李俊, 廖强, 等. 2009. 过硫酸钾为电子受体的微生物燃料电池性能特性[J]. 工程热物理学报, 2009, 30(8): 1396–1398.
DOI:10.3321/j.issn:0253-231X.2009.08.039 |
Ghauch A, Tuqan A M, Kibbi N. 2012. Ibuprofen removal by heated persulfate in aqueous solution:A kinetics study[J]. Chemical Engineering Journal, 197: 483–492.
DOI:10.1016/j.cej.2012.05.051
|
Gao H P, Chen J B, Zhang Y L, et al. 2016. Sulfate radicals induced degradation of Triclosan in thermally activated persulfate system[J]. Chemical Engineering Journal, 306: 522–530.
DOI:10.1016/j.cej.2016.07.080
|
高乃云, 朱延平, 谈超群, 等. 2013. 热活化过硫酸盐氧化降解敌草隆[J]. 华南理工大学学报(自然科学版), 2013, 41(12): 36–42.
DOI:10.3969/j.issn.1000-565X.2013.12.007 |
孔令国, 王玲, 薛建国, 等. 2010. 负载型三维粒子电极降解甲基橙模拟废水研究[J]. 中国环境科学, 2010, 30(4): 516–521.
|
孔晓英, 李连华, 马隆龙, 等. 2012. 不同膜微生物燃料电池的性能[J]. 太阳能学报, 2012, 33(5): 882–885.
DOI:10.3969/j.issn.0254-0096.2012.05.030 |
Lin H, Wu J, Zhang H. 2013. Degradation of bisphenol A in aqueous solution by a novel electro/Fe3+/peroxydisulfate process[J]. Separation & Purification Technology, 117(4): 18–23.
|
骆海萍, 刘广立, 张仁铎, 等. 2008. 以苯酚为燃料的微生物燃料电池产电特性[J]. 环境科学学报, 2008, 28(7): 1279–1283.
DOI:10.3321/j.issn:0253-2468.2008.07.003 |
Li J, Fu Q, Liao Q, et al. 2009. Persulfate:A self-activated cathodic electron acceptor for microbial fuel cells[J]. Journal of Power Sources, 194(1): 269–274.
DOI:10.1016/j.jpowsour.2009.04.055
|
Liu J L, Zhong S, Song Y P, et al. 2018. Degradation of tetracycline hydrochloride by electro-activated persulfate oxidation[J]. Journal of Electroanalytical Chemistry, 809: 74–79.
DOI:10.1016/j.jelechem.2017.12.033
|
Liu L, LI F B, Feng C H, et al. 2009. Microbial fuel cell with an azo-dye-feeding cathode[J]. Applied Microbiology and Biotechnology, 85: 175–183.
DOI:10.1007/s00253-009-2147-9
|
李明玉, 尚薇, 李心乐, 等. 2009. 光电化学协同催化降解甲基橙的研究[J]. 中国环境科学, 2009, 29(5): 512–517.
DOI:10.3321/j.issn:1000-6923.2009.05.012 |
梁鹏, 范明志, 曹效鑫, 等. 2007. 微生物燃料电池表观内阻的构成和测量[J]. 环境科学, 2007, 28(8): 1894–1898.
DOI:10.3321/j.issn:0250-3301.2007.08.043 |
李霞.2017.还原铁粉和活性炭活化过硫酸钠处理甲基橙废水的试验研究[D].郑州: 郑州大学
http://cdmd.cnki.com.cn/Article/CDMD-10459-1017129028.htm |
牟姝君, 袁李秀, 任月萍, 等. 2014. 铜离子对双室微生物燃料电池电能输出的影响研究[J]. 环境科学, 2014, 35(7): 2791–2797.
|
Miran W, Jang J, Nawaz M, et al. 2017. Mixed sulfate-reducing bacteria-enriched microbial fuel cells for the treatment of wastewater containing copper[J]. Chemosphere, 189: 134–142.
DOI:10.1016/j.chemosphere.2017.09.048
|
Nie M H, Yan C X, Li M, et al. 2015. Degradation of chloramphenicol by persulfate activated by Fe2+ and zerovalent iron[J]. Chemical Engineering Journal, 279: 507–515.
DOI:10.1016/j.cej.2015.05.055
|
Richard J W, Mushtaque A, Amanda K H, et al. 2018. Persulfate activation by glucose for in situ chemical oxidation[J]. Water research, 133: 247–254.
DOI:10.1016/j.watres.2018.01.050
|
唐琪, 王玉如, 郭菁豪, 等. 2017. CuO活化过硫酸盐对孔雀石绿的降解[J]. 环境工程学报, 2017, 11(4): 2084–2090.
|
Wang C W, Liang C J. 2015. Oxidative degradation of TMAH solution with UV persulfate activation[J]. Chemical Engineering Journal, 254: 472–478.
|
Wu Y L, Romian P, Marcello B, et al. 2017. Activation of persulfate by Fe(Ⅲ) species:Implications for 4-tert-butylphenol degradation[J]. Journal of Hazardous Materials, 322: 380–386.
DOI:10.1016/j.jhazmat.2016.10.013
|
尤世界, 赵庆良, 姜瑶秋, 等. 2006. 废水同步生物处理与生物燃料电池发电研究[J]. 环境科学, 2006, 27(9): 1786–1790.
DOI:10.3321/j.issn:0250-3301.2006.09.016 |
Zhang H, Wang Z, Liu C, et al. 2014. Removal of COD from landfill leachate by an electro/Fe2+/peroxydisulfate process[J]. Chemical Engineering Journal, 250: 76–82.
DOI:10.1016/j.cej.2014.03.114
|
张剑桥.2016.Cu2+强化Fe2+活化过硫酸盐降解苯酚的效能与机理研究[D].哈尔滨: 哈尔滨工业大学
http://cdmd.cnki.com.cn/Article/CDMD-10213-1016739858.htm |
郑垒, 汪晓军, 汪星志, 等. 2016. 过硫酸盐-石灰苏打处理印染反渗透浓水研究[J]. 环境科学学报, 2016, 36(1): 166–171.
|
朱美月, 刘维平. 2017. 铜离子为电子受体的MFC产电性能及废水处理[J]. 工业水处理, 2017, 37(12): 64–67.
DOI:10.11894/1005-829x.2017.37(12).064 |