 2019, Vol. 39
2019, Vol. 39



2. 中国科学院生态环境研究中心, 饮用水科学与技术重点实验室, 北京 100085;
3. 武汉纺织大学, 环境工程学院, 武汉 430200
2. Key Laboratory of Drinking Water Science and Technology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085;
3. School of Environmental Engineering, Wuhan Textile University, Wuhan 430200
饮用水消毒是20世纪公共卫生的主要进步之一, 然而自20世纪70年代以来, 人们已经认识到氯化消毒剂可以与水中的天然有机物(NOM)反应形成消毒副产物(DBPs)(Rook, 1974; Krasner et al., 1989; Singer, 1994), 常见的DBPs, 如三卤甲烷(THMs)、卤乙酸(HAAs)、溴酸盐, 以及“新兴”DBPs等引起了人们的广泛关注(Richardson et al., 2007).饮用水中较常检出的THMs共有4种, 即三氯甲烷(TCM)、二氯一溴甲烷(BDCM)、一氯二溴甲烷(DBCM)和三溴甲烷(TBM), 当原水中不含溴离子时, TCM为主要成分, 此外, TCM会通过非遗传毒性作用诱导动物产生肿瘤, 致癌机制包括再生细胞分化和细胞毒性(赵玉丽等, 2011).卤乙腈(HANs)作为一类新兴含氮类DBPs, 其细胞毒性和基因毒性比HAAs高出两个数量级, 其中二氯乙腈(DCAN)已被确认为具有致畸、致癌、致突变性(Ahmed et al., 2000; Yu et al., 2015), 且卤代乙腈中二氯乙腈生成量通常最高.DBPs会直接影响饮用水的感官性状, 增加水体的嗅、味, 部分DBPs具有“三致”作用, 长期饮用氯化消毒水与消化及泌尿系统癌变风险呈正相关性(Nieuwenhuijsen et al., 2000; 赵玉丽等, 2011).流行病学研究结果表明慢性暴露于DBPs下, 患上膀胱癌的风险会增加, 对健康产生不良影响(Villanueva et al., 2006; Hrudey et al., 2015).
NOM是自然界广泛存在的大分子有机物, 由动植物在循环过程中腐烂分解所成, 腐殖酸(HA)和富里酸(FA)是NOM的主要成分.与HA相比, FA具有较小的分子量和更多的含氧官能团(Weng et al., 2006), 且FA的溶解性和移动性较强, 对某些土壤的淋溶和淀积有很大作用.NOM具有复杂的有机结构和活性基团, 会与水中的其它有机污染物发生反应, 或与金属离子发生络合、螯合等反应, 不仅干扰污染物的有效去除而且容易生成毒性更大的副产物(Widrig et al., 1996; Lin et al., 2000; Korshin et al., 2002; White et al., 2003; Roccaro et al., 2008).为了探究NOM的特性与DBPs生成势之间的关系, 一些替代参数已广泛用于量化NOM反应性在DBPs形成中的作用(Beauchamp et al., 2018), 例如A254(NOM在254 nm处的吸光度)和SUVA254(特征紫外吸光度, 即A254与溶解性有机碳(DOC)的比值).根据SUVA254可初步比较水中有机物的芳香化程度和分子量大小(Roccaro et al., 2009).Korshin和Roccaro等研究发现基于差分吸收光谱272 nm处的吸光度(-A272)与总有机卤素(TOX)和不同氯剂量、反应时间和温度下DBPs的形成具有很强的相关性.不同的水源, NOM的性质也存在较大的差异, 因此需找到有效的手段对NOM的特性进行分析, 进而将其去除以改善水质.常用的NOM的表征技术有:DOC的测定、分子量分布、吸光度以及三维荧光光谱(3D-EEM).
水处理过程中通常采用化学预氧化技术对DBPs的前驱物如NOM进行控制.化学预氧化可降低胶体表面静电斥力, 促进胶体脱稳, 提高混凝效果, 从而有助于NOM的去除.高锰酸钾(PM)于1659年被Glauber发现, 并在20世纪初用于水处理过程.作为一种传统的预氧化剂, 其储存运输简单安全, 在氧化降解有机物过程中不会产生有毒、有害的卤代副产物, 其最终还原产物为环境友好难溶于水的MnO2, 可通过重力作用沉淀分离, 且MnO2还可以通过吸附、氧化、助凝等作用与PM协同去除污染物(Zhao et al., 2018).但PM氧化作用较温和, 针对污染物的氧化速率较慢而且无法去除难降解有机污染物.此外, PM的水溶液为紫红色, 实际应用中为了避免水体色度的增加, 投加浓度会比较低.为了提高对有机物的吸附及氧化能力, 目前已将PM与其它药剂联合使用, 通过复合药剂的协同作用强化污染物的去除.近年来, 为了提高PM对某些污染物氧化能力, 诸多关于PM氧化污染物的动力学研究相继展开(Guan et al., 2010; Mahmoodlu et al., 2014).孙波等在探究PM氧化动力学的过程中发现, 在投加一定浓度的亚硫酸氢钠(BS)作为PM的猝灭剂时不但没有终止PM氧化污染物, 反而发现污染物的浓度瞬间降低, 之后通过时间分辨三维紫外光谱、电子顺磁共振波谱、络合剂影响以及pH变化等实验证实BS活化PM原位生成了水解态的Mn(Ⅲ).研究发现PM/BS体系可在毫秒级时间尺度内降解有机污染物, 且氧化能力极强, 能够去除单独PM无法氧化的有机污染物, 其氧化速率是传统方法的几万到上百万倍(Sun et al., 2015), 然而PM/BS氧化工艺对水中NOM的影响尚未有报道.PM/BS体系主要是通过产生Mn(Ⅲ)及氧硫自由基等强氧化性中间产物对有机物如FA进行降解, 其氧化大量有机污染物所需的接触时间极短, 有望在水处理过程中强化降解微量污染物.PM/BS体系所需的试剂对于工程规模较为实用的, 其形成的最终产物是SO42-, Mn2+和MnO2, 锰的产物都可以在常规水处理工艺中得到进一步控制(Sun et al., 2015; Sun et al., 2016; Gao et al., 2017).PM/BS体系反应的主要过程如下所示:
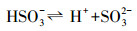
|
(1) |
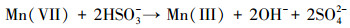
|
(2) |

|
(3) |

|
(4) |
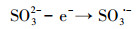
|
(5) |
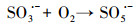
|
(6) |
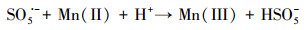
|
(7) |
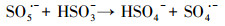
|
(8) |
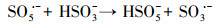
|
(9) |
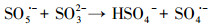
|
(10) |
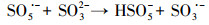
|
(11) |
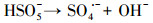
|
(12) |
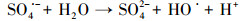
|
(13) |

|
(14) |
单独PM、PM/BS体系在氧化微量有机物的同时会不可避免的影响NOM结构, 从而影响DBPs的生成.因此, 本文考察了3种含碳类消毒副产物(C-DBPs)(1, 1-二氯丙酮(1, 1-DCP)、1, 1, 1-三氯丙酮(1, 1, 1-TCP)、三氯甲烷(TCM))和3种含氮类消毒副产物(N-DBPs)(三氯乙腈(TCAN)、二氯乙腈(DCAN)、三氯硝基甲烷(TCNM))的生成势.选取FA为NOM的代表, 研究了PM/BS体系在预氧化过程中对NOM结构及去除的影响.主要内容包括以下两个部分:①评估单独PM、PM/BS体系在氧化过程中对FA的总有机碳(TOC)、254 nm处的吸光度(UV254)、3D-EEM等指标的变化.②考察单独PM氧化、PM/BS氧化FA前后, 其DBPs生成势的变化, 从而探究PM/BS预氧化影响NOM结构特征及DBPs生成势的机理.
2 实验材料与方法(Material and methods) 2.1 化学试剂与仪器 2.1.1 化学试剂高锰酸钾、十二水合磷酸氢二钠、十二水合磷酸二氢钠、十二水合磷酸二氢钾、乙二胺四乙酸二钠盐、无水DPD硫酸盐、次氯酸钠、硫酸、氢氧化钠、无水硫酸钠均由国药集团化学试剂有限公司提供, 高锰酸钾为优级纯, 其他为分析纯; 亚硫酸氢钠为分析纯, 由北京北化精细化学品有限责任公司提供; 甲基叔丁基醚由北京市百灵威科技有限公司提供(纯度99%); 黄腐酸(FA)由上海麦克林生化科技有限公司提供(纯度85%).FA属于小分子有机物, 分子量在几百到几千, 分子结构中含有的C、O元素较多, 酸性官能团的含量也较高, 易溶于水(Schulten, 1995), 本实验采用超纯水直接溶解方式配制FA, 浓度为1.0 g · L-1.
2.1.2 实验仪器AL104电子天平(梅特勒-托利多仪器有限公司); 84-1A磁力搅拌器(上海司乐仪器有限公司); KQ-300DE超声波清洗仪(昆山市超声仪器有限公司); FiveEasy实验室pH计(梅特勒-托利多); Milli-Q A10超纯水仪(默克密理博); DR6000紫外可见分光光度计(哈希公司); DR5000紫外可见分光光度计(哈希公司); VX-Ⅲ振荡机(TARGIN); 7890A气相色谱仪(安捷伦科技有限公司); TOC-Vcph TOC分析仪(SHIMADZU); CARY ECLIPSE荧光分光光度计(安捷伦科技有限公司).
2.2 实验方法预氧化实验方法:将BS和PM先后加入到含有FA(3.0 mg · L-1)的水溶液中, 其中PM和BS的摩尔比采用孙波等发现的最佳摩尔比1 : 5(Sun et al., 2015).同时设置单独FA, FA和PM作为对照.将反应液置于磁力搅拌器上搅拌30 min, 然后将反应液转移至40 mL棕色瓶中.按比例1 : 5设置不同浓度的PM及BS预氧化FA, 探究PM及BS浓度对DBPs生成势的影响; 分别设置反应pH为5.5、6.5、7、7.5、8.5, 探究pH对DBPs生成势的影响.所有实验均在室温条件下进行, 如无特殊说明pH均控制在6.5(缓冲).
DBPs生成势实验方法:将次氯酸钠储备液加入到上述反应液中, 氯的投量为20 mg · L-1.5 min后采用DPD法测定PM和PM/BS体系中的氯浓度, 补充适量的氯到PM/BS体系, 摇匀并置于暗处暗反应24 h.
2.3 分析测试方法DBPs生成分析:取20 mL暗反应24 h后的反应液于40 mL玻璃瓶中, 加入6 g无水硫酸钠, 然后加入2 mL甲基叔丁基醚(MTBE).将上述样品置于振荡机上振荡10 min, 振荡完毕后静置10 min, 待有机相与水相分层后, 萃取上层有机相, 过0.45 μm滤膜于棕色色谱瓶中以待后续分析.
样品经液液萃取后提取上层有机相, 通过GC-ECD进行分析(安捷伦7890A气相色谱仪, 电子捕获检测器).进样口采用分流模式, 分流比为20 : 1.所用色谱柱为安捷伦19091J-413型号色谱柱, 基本参数为:325 ℃、30 m×320 μm×0.25 μm, 流速为1 mL · min-1, 加热器温度为200 ℃, 压力设定为4.7282 psi, 平均流速为18.689 cm · s-1, 隔垫吹扫流量为2 mL · min-1, 滞留时间为2.6754 min.电子捕获器, 检测器温度为300 ℃, 进样口温度为200 ℃.程序升温:40 ℃, 保持5 min, 以1 ℃ · min-1升至45 ℃(保持3 min), 以10 ℃ · min-1升至135 ℃后, 再以25 ℃ · min-1升至220 ℃.进样体积1 μL.
采用TOC-Vcph TOC分析仪测定TOC含量.采用DR 5000紫外分光光度计测得UV-Vis吸收光谱(波长扫描范围为200~800 nm).3D-EEM采用CARY ECLIPSE荧光分光光度计测定, 以氙灯为激发光源; 以超纯水作空白校正水样的拉曼散射.扫描间隔为Ex=5 nm、Em=5 nm; 光谱仪扫描范围为激发波长/发射波范围为220~420 nm/280~550 nm; 响应时间为自动.采用N, N-二乙基对苯二胺分光光度法对余氯量进行测定.
3 结果与讨论(Results and discussion) 3.1 NOM的特性表征 3.1.1 FA经预氧化后紫外可见吸收光谱的变化紫外可见吸收光谱是一种相对简单的表征NOM的方法, 不需制备特定的样品且可显示有关NOM发色基团的相关信息(Huang et al., 2018).图 1为不同投加量下PM和PM/BS体系预氧化FA(3.0 mg · L-1)30 min的UV-Vis图谱特征峰.通过之前的研究得知PM在525 nm处存在特征峰(ε=2500 M-1 · cm-1)(Zhao et al., 2016).由图可知, 在单独PM预氧化体系中, 增大PM的浓度至250 μmol · L-1, 525 nm处PM的特征峰存在, 通过计算得出30 min后PM的浓度降低至222.8 μmol · L-1, 但PM/BS(250 μmol · L-1 : 1250 μmol · L-1)体系中PM的特征峰几乎消失, 计算得出30 min后PM的浓度降低至21.2 μmol · L-1, 这说明PM/BS体系具有较强的氧化活性.如图 1a所示, FA在240~280 nm之间存在吸收峰, 表明FA中存在C═C双键结构.在所有体系中, 随着预氧化剂投加量的增加, 吸光度值均呈增大的趋势.这可能是因为在高剂量预氧化剂的作用下, FA的结构被破坏, 转化成了一些小分子物质, 各官能团之间的干扰变弱, 因而发色基团能够吸收更多的光强从而产生更大的吸光度.
 |
| 图 1 PM与PM/BS系统不同投加量下紫外可见光吸收图谱 (a.PM/FA体系; b.PM/BA/FA体系) Fig. 1 UV-Vis absorption spectra of PM and PM/BS systems at different dosages (a. PM/FA system; b. PM/BS/FA system) |
图 2考察了PM与BS不同投加量的情况下FA的TOC的去除情况及SUVA254的变化, 结果表明, TOC的去除率随着PM与BS浓度的增大总体呈上升的趋势, 这可能是因为当氧化剂浓度增大时, 反应产生了更多的中间活性物质, 例如Mn(Ⅲ)、氧硫自由基等, 这些中间活性物质具有极强的氧化性(Sun et al., 2018), 从而使有机物矿化为无机物的比例增大.值得注意的是, 在PM/BS体系中, 虽然TOC去除率随着投加量的增大有所升高, 但在最大投加量的条件下也只达到了12%左右, 此结果表明FA较难被矿化.此外, 由于FA是易溶于水的小分子有机物, 难以被PM/BS体系中产生的MnO2吸附, 从而导致TOC去除率较低.
 |
| 图 2 PM/BS不同投加量下TOC的去除率以及SUVA254变化图 Fig. 2 TOC removal and SUVA254 variation for different PM/BS dosages |
因为碳原子和有机结构之间形成的双键在254 nm处有吸收峰, 所以常用UV254来表征NOM的芳香性(Marais et al., 2017).如图 2所示, FA的SUVA254值均随投加量的增大而增加.结合TOC去除率数据, 当TOC去除率升高时, UV254值原理上说会相应的减小, 但SUVA254值却不断升高.这可能是因为随着投加量的增大, 水体的色度也会相应的加深, 从而影响SUVA254值的测定.另一方面, SUVA254升高表明水体中不饱和类有机物浓度增加, 这说明在PM与BS预氧化的作用下, 胶体的稳定性会遭到破坏, 部分有机物会释放出来.
3.1.3 FA经预氧化及加氯暗反应后的3D-EEM分析三维荧光近来被广泛用于NOM的表征, 其光谱特征可反应分子结构以及分子内部、分子间相互作用的动力学等信息.图 3为3 mg · L-1的FA经PM(50 μmol · L-1)与PM/BS(50 μmol · L-1 : 250 μmol · L-1)体系预氧化及氯化反应24 h后的荧光光谱图.Henderson等研究表明, 荧光光谱根据荧光峰的位置可大致分为5个区域, 分别代表 5种不同类别的NOM(Henderson et al., 2009).区域Ⅰ和区域Ⅱ的位置分别是在Ex/Em=200~250 nm/280~320 nm、Ex/Em=200~250 nm/320~380 nm, 主要代表了芳香性蛋白类物质; 区域Ⅲ的位置在Ex/Em=200~250 nm /380~500 nm, 代表了FA类物质; 区域Ⅳ位置在Ex/Em=250~280 nm/280~380 nm, 代表了溶解性微生物产物, 区域Ⅴ的位置在Ex/Em=250~400 nm/380~500 nm, 代表了HA类物质.通过图 3a中FA的荧光峰分布位置可知, FA中含有芳香性蛋白类物质.由图 3b可以看出FA的荧光光谱发生明显的变化, 经过PM预氧化之后FA的峰由Ex/Em=230 nm/390 nm右移至Ex/Em= 210 nm/450 nm, 最大荧光强度由80 a.u.降低至40 a.u., 发光范围也有了很大的缩小.由图 3c可知, 在PM/BS预氧化后FA的峰右移至Ex/Em=210 nm/450 nm, 最大荧光强度由80 a.u.降低至40 a.u., 这与PM体系基本相似, 而发光范围进一步缩小, 这表明PM/BS体系产生的中间氧化剂可有效破坏FA的部分发色基团.FA有机物结构的变化将有可能影响其DBPs生成势, 需进一步深入探究.
 |
| 图 3 FA预氧化30 min及加氯暗反应24 h后的3D-EEM图谱 (a.空白; b.PM系统预氧化及加氯暗反应; c.PM/BS系统预氧化及加氯暗反应) Fig. 3 3D-EEM after FA pre-oxidation for 30 min and chlorination for 24 h (a. blank; b. PM system pre-oxidation and chlorination; c. PM/BS system pre-oxidation and chlorination) |
PM的投加量对DBPs生成势的影响如图 4所示.由图可知, 在检测的6种DBPs中, TCM的生成浓度显著高于其它5种DBPs, 其浓度大约是其它C-DBPs和N-DBPs的20~500倍.随着PM投加量的增大, TCM的浓度先升高后降低.在PM投加量为20 μmol · L-1时, TCM的浓度达到270.9 μg · L-1, 继续增大PM投加量TCM的浓度逐渐降低, 结合上述3D-EEM的现象得知单独PM能有效氧化FA, 这可能是造成TCM浓度逐渐降低的原因.总体来看, PM预氧化时, N-DBPs的生成势较小, 且随着PM投加量的增大, 其浓度总体呈逐渐降低趋势, 这可能是由于在预氧化过程中, 生成的N-DBPs不稳定, 会发生水解从而浓度下降.由图 4可知, 在PM/BS体系中, TCM的量呈先下降后上升的趋势, 在PM投加量为50 μmol · L-1, BS投加量为250 μmol · L-1时, 体系中TCM浓度仅为123.7 μg · L-1.与PM体系相比, PM/BS体系中, N-DBPs的生成势显著增大, 且DCAN的浓度随着投加量的增大而逐渐升高, 这与PM体系中的变化恰好相反.对比空白对照组, PM体系以及PM/BS体系能够更直观的发现两种体系中DBPs生成势的差异.在不投加PM和BS时, TCM浓度约为161.8 μg · L-1, DCAN浓度约为8.7 μg · L-1; 在PM体系中, 当PM投加量为100 μmol · L-1时TCM浓度增加至约207.7 μg · L-1, DCAN浓度降低至约2.9 μg · L-1; 而在PM/BS体系中, 当PM投加量为100 μmol · L-1, BS投加量为500 μmol · L-1时, TCM浓度增加至约186.3 μg · L-1, DCAN浓度增加至约14.8 μg · L-1.
 |
| 图 4 PM及PM/BS投加量对C-DBPs和N-DBPs生成势的影响 (a.PM系统中产生的C-DBPs; b.PM/BS系统中产生的C-DBPs; c.PM系统中产生的N-DBPs; d.PM/BS系统中产生的N-DBPs) Fig. 4 Effect of PM and PM/BS dosage on the generation potential of C-DBPs and N-DBPs (a. C-DBPs generated in PM system; b. C-DBPs generated in PM/BS system; c. N-DBPs generated in PM system; d. N-DBPs generated in PM/BS system) |
溶液的pH值会影响NOM的存在状态, 同时会影响PM、PM/BS对DBPs的氧化过程.如图 5所示, 在无预氧化和PM预氧化的条件下, TCM的浓度随pH的升高而增大, 这与Nihemaiti等研究得出的结论一致(Nihemaiti et al., 2016).pH会影响不稳定DBPs的水解速率, 在碱性条件下, 其它不稳定的DBPs水解转化为TCM的量增多(李林林等, 2016).在无预氧化的条件下, 1, 1, 1-TCP浓度随pH的上升而逐渐减小, 但N-DBPs浓度随pH的上升而逐渐增加, 这可能是因为FA含有较多的酸性官能团, 导致溶液呈酸性, 在预氧化过程中会影响DBPs的水解(李林林等, 2016).pH对PM的反应动力学及氧化途径有显著的影响.在酸性条件下, PM具有强氧化性, 随着pH的逐渐升高, 氧化能力逐渐减弱, 在碱性条件下, 不能将有机物完全氧化, 而是会促进胶体脱稳, 增进混凝过程中有机物的去除.如图 5c和5d所示, 在PM预氧化后, 随着pH的增大, 1, 1-DCP和1, 1, 1-TCP浓度均呈下降趋势, 但TCM的浓度呈现上升趋势, 这与之前的研究结果相吻合(Hansen et al., 2013; 李林林等, 2016).如图 5e和5f所示, 在PM/BS体系中, C-DBPs及N-DBPs总浓度随pH的增大呈现先降低后升高后降低的趋势, 在pH为7.5时生成势达到最大值.在pH为7.5时, 无预氧化的条件下生成的TCM浓度约为100.7 μg · L-1, DCAN浓度约为7.5 μg · L-1; 在PM预氧化条件下二者浓度分别约为127.5 μg · L-1和9.7 μg · L-1; 而在PM/BS预氧化条件下生成的TCM浓度高达217.1 μg · L-1, DCAN浓度约为9.3 μg · L-1.结合图 5无预氧化条件下的结果得知单独FA的DBPs生成势随着pH增大而增大, 此外, 由于碱性条件HSO32-更易转化为SO32-以及Mn(Ⅲ)更高的歧化率, 因此PM/BS体系在酸性下有更好的氧化活性(Tartar et al., 1941; Sun et al., 2015).在pH 7.5时DBPs生成量最高, 而实际水体的pH范围多在7.5左右, PM/BS氧化NOM可能引发的DBPs的提升需格外重视.
 |
| 图 5 无预氧化, PM预氧化及PM/BS预氧化条件下, pH对C-DBPs及N-DBPs生成势的影响 ((a)、(c)、(e)分别为无预氧化, PM预氧化及PM/BS预氧化条件下pH对C-DBPs生成势的影响; (b)、(d)、(f)分别为无预氧化, PM预氧化及PM/BS预氧化条件下pH对N-DBPs生成势的影响) Fig. 5 Effect of pH on the formation potential of C-DBPs and N-DBPs under without pre-oxidation, PM pre-oxidation and PM/BS pre-oxidation conditions. ((a), (c), (e) are the effect of pH on the formation potential of C-DBPs under without pre-oxidation, PM pre-oxidation and PM/BS pre-oxidation conditions; (b), (d), (f) are the effect of pH on the formation potential of N-DBPs under without pre-oxidation, PM pre-oxidation and PM/BS pre-oxidation conditions) |
1) PM/BS体系可改变FA的结构, 将大分子芳香族官能团、结构转化为小分子有机物.随着PM与BS投加量的增大, TOC去除率和SUVA254会随之增大.
2) PM/BS预氧化FA不能显著降低C-DBPs及N-DBPs的浓度, 与PM单独氧化相比, 1, 1, 1-TCP, 1, 1-DCP及DCAN的浓度会升高.随着投加量的增大, C-DBPs的总浓度呈先降低后升高的趋势, 而N-DBPs的总浓度则显著上升.
3) pH对FA的DBPs生成势有显著影响, 在无预氧化和PM氧化体系中, 除TCM外其他C-DBPs浓度随pH的增大而下降.在PM/BS体系中, C-DBPs及N-DBPs总浓度总体呈先降低后升高再降低的趋势, pH 7.5时达到最大值.在pH为7.5时, 无预氧化的条件下生成的TCM浓度约为100.7 μg · L-1, DCAN浓度约为7.5 μg · L-1; 在PM预氧化条件下二者浓度分别约为127.5 μg · L-1和9.7 μg · L-1; 而在PM/BS预氧化条件下生成的TCM浓度高达217.1 μg · L-1, DCAN浓度约为9.3 μg · L-1.
Ahmed A E, Aronson J, Jacob S. 2000. Induction of oxidative stress and TNF-α secretion by Dichloroacetonitrile, a water disinfectant by-product, as possible mediators of apoptosis or necrosis in a murine macrophage cell line (RAW)[J]. Toxicology in Vitro, 14(3): 199–210.
DOI:10.1016/S0887-2333(00)00019-9
|
Beauchamp N, Laflamme O, Simard S, et al. 2018. Relationships between DBP concentrations and differential UV absorbance in full-scale conditions[J]. Water Research, 131: 110–121.
DOI:10.1016/j.watres.2017.12.031
|
Guan X, He D, Ma J, et al. 2010. Application of permanganate in the oxidation of micropollutants:a mini review[J]. Frontiers of Environmental Science & Engineering in China, 4(4): 405–413.
|
Gao Y, Jiang J, Zhou Y, et al. 2017. Unrecognized role of bisulfite as Mn (Ⅲ) stabilizing agent in activating permanganate (Mn (Ⅶ)) for enhanced degradation of organic contaminants[J]. Chemical Engineering Journal, 327: 418–422.
DOI:10.1016/j.cej.2017.06.056
|
Hrudey S E, Backer L C, Humpage A R, et al. 2015. Evaluating evidence for association of human bladder cancer with drinking-water chlorination disinfection by-products[J]. Journal of Toxicology and Environmental Health, Part B, 18(5): 213–241.
DOI:10.1080/10937404.2015.1067661
|
Henderson R K, Baker A, Murphy K R, et al. 2009. Fluorescence as a potential monitoring tool for recycled water systems:A review[J]. Water Research, 43(4): 863–881.
DOI:10.1016/j.watres.2008.11.027
|
Huang S, Gan W, Yan M, et al. 2018. Differential UV-vis absorbance can characterize the reaction of organic matter with ClO2[J]. Water Research, 139: 442–449.
DOI:10.1016/j.watres.2018.04.006
|
Hansen K, Albrechtsen H J, Andersen H R. 2013. Optimal pH in chlorinated swimming pools-balancing formation of by-products[J]. Journal of Water and Health, 11(3): 465–472.
DOI:10.2166/wh.2013.156
|
Krasner S W, McGuire M J, Jacangelo J G, et al. 1989. The occurrence of disinfection by-products in US drinking water[J]. Journal-American Water Works Association, 81(8): 41–53.
DOI:10.1002/j.1551-8833.1989.tb03258.x
|
Korshin G V, Wu W W, Benjamin M M, et al. 2002. Correlations between differential absorbance and the formation of individual DBPs[J]. Water Research, 36(13): 3273–3282.
DOI:10.1016/S0043-1354(02)00042-8
|
Lin C F, Lin T Y, Hao O J. 2000. Effects of humic substance characteristics on UF performance[J]. Water Research, 34(4): 1097–1106.
DOI:10.1016/S0043-1354(99)00273-0
|
李林林, 刘佳蒙, 宋弼尧, 等. 2016. 饮用水中典型微生物消毒过程中消毒副产物的生成规律[J]. 中国环境科学, 2016, 36(12): 3631–3638.
DOI:10.3969/j.issn.1000-6923.2016.12.014 |
Mahmoodlu M G, Hassanizadeh S M, Hartog N. 2014. Evaluation of the kinetic oxidation of aqueous volatile organic compounds by permanganate[J]. Science of the Total Environment, 485: 755–763.
|
Marais S S, Ncube E J, Msagati T A M, et al. 2017. Investigation of natural organic matter (NOM) character and its removal in a chlorinated and chloraminated system at Rand Water, South Africa[J]. Water Science and Technology:Water Supply, 17(5): 1287–1297.
DOI:10.2166/ws.2017.028
|
Nieuwenhuijsen M J, Toledano M B, Eaton N E, et al. 2000. Chlorination disinfection byproducts in water and their association with adverse reproductive outcomes:a review[J]. Occupational and Environmental Medicine, 57(2): 73–85.
DOI:10.1136/oem.57.2.73
|
Nihemaiti M, Le Roux J, Hoppe-Jones C, et al. 2016. Formation of haloacetonitriles, haloacetamides, and nitrogenous heterocyclic byproducts by chloramination of phenolic compounds[J]. Environmental Science & Technology, 51(1): 655–663.
|
Rook J J. 1974. Formation of haloforms during chlorination of natural waters[J]. Water Treat Exam, 23: 234–243.
|
Richardson S D, Plewa M J, Wagner E D, et al. 2007. Occurrence, genotoxicity, and carcinogenicity of regulated and emerging disinfection by-products in drinking water:a review and roadmap for research[J]. Mutation Research/Reviews in Mutation Research, 636(1/3): 178–242.
|
Roccaro P, Chang H S, Vagliasindi F G A, et al. 2008. Differential absorbance study of effects of temperature on chlorine consumption and formation of disinfection by-products in chlorinated water[J]. Water Research, 42(8/9): 1879–1888.
|
Roccaro P, Vagliasindi F G A. 2009. Differential vs.absolute UV absorbance approaches in studying NOM reactivity in DBPs formation:Comparison and applicability[J]. Water Research, 43(3): 744–750.
DOI:10.1016/j.watres.2008.11.007
|
Singer P C. 1994. Control of disinfection by-products in drinking water[J]. Journal of Environmental Engineering, 120(4): 727–744.
DOI:10.1061/(ASCE)0733-9372(1994)120:4(727)
|
Sun B, Guan X, Fang J, et al. 2015. Activation of manganese oxidants with bisulfite for enhanced oxidation of organic contaminants:the involvement of Mn (Ⅲ)[J]. Environmental Science & Technology, 49(20): 12414–12421.
|
Sun B, Dong H, He D, et al. 2016. Modeling the kinetics of contaminants oxidation and the generation of manganese (Ⅲ) in the permanganate/bisulfite process[J]. Environmental Science & Technology, 50(3): 1473–1482.
|
Schulten H R. 1995. The three-dimensional structure of humic substances and soil organic matter studied by computational analytical chemistry[J]. Fresenius' Journal of Analytical Chemistry, 351(1): 62–73.
DOI:10.1007/BF00324293
|
Sun B, Bao Q, Guan X. 2018. Critical role of oxygen for rapid degradation of organic contaminants in permanganate/bisulfite process[J]. Journal of Hazardous Materials, 352: 157–164.
DOI:10.1016/j.jhazmat.2018.03.024
|
Tartar H V, Garretson H H. 1941. The thermodynamic ionization constants of sulfurous acid at 25[J]. Journal of the American Chemical Society, 63(3): 808–816.
DOI:10.1021/ja01848a049
|
Villanueva C M, Cantor K P, Grimalt J O, et al. 2006. Bladder cancer and exposure to water disinfection by-products through ingestion, bathing, showering, and swimming in pools[J]. American Journal of Epidemiology, 165(2): 148–156.
DOI:10.1093/aje/kwj364
|
Weng L, Van Riemsdijk W H, Koopal L K, et al. 2006. Adsorption of humic substances on goethite:comparison between humic acids and fulvic acids[J]. Environmental Science & Technology, 40(24): 7494–7500.
|
Widrig D L, Gray K A, McAuliffe K S. 1996. Removal of algal-derived organic material by preozonation and coagulation:monitoring changes in organic quality by pyrolysis-GC-MS[J]. Water Research, 30(11): 2621–2632.
DOI:10.1016/S0043-1354(96)00162-5
|
White D M, Garland D S, Narr J, et al. 2003. Natural organic matter and DBP formation potential in Alaskan water supplies[J]. Water Research, 37(4): 939–947.
DOI:10.1016/S0043-1354(02)00425-6
|
Yu Y, Reckhow D A. 2015. Kinetic analysis of haloacetonitrile stability in drinking waters[J]. Environmental Science & Technology, 49(18): 11028–11036.
|
赵玉丽, 李杏放. 2011. 饮用水消毒副产物:化学特征与毒性[J]. 环境化学, 2011, 30(1): 20–33.
|
Zhao H, Wang L, Zhang H, et al. 2018. Effect of potassium permanganate dosing position on the performance of coagulation/ultrafiltration combined process[J]. Chinese Journal of Chemical Engineering, 26(1): 89–95.
DOI:10.1016/j.cjche.2017.03.037
|
Zhao X, Salhi E, Liu H, et al. 2016. Kinetic and mechanistic aspects of the reactions of iodide and hypoiodous acid with permanganate:Oxidation and disproportionation[J]. Environmental Science & Technology, 50(8): 4358–4365.
|