 2019, Vol. 39
2019, Vol. 39



2. 南京大学环境规划设计研究院股份公司, 南京 210093
2. Academy of Environmental Planning & Design, Co., Ltd., Nanjing University, Nanjing 210093
水体中类金属污染物无机砷因会造成器官致癌, 黑足病以及慢性中毒等严重危害人类健康作用而被广泛关注(Naidu et al., 2006; Liu et al., 2010).虽然世界卫生组织对水体中砷浓度标准有明确的规定(10 μg·L-1), 但在孟加拉国、阿根廷、墨西哥等多个国家和地区仍然存在由于地表水或地下水中高浓度无机砷在而引发的环境污染事件(Matschullat, 2000; Mandal et al., 2002; Smedley et al., 2002; Anawar et al., 2003; Tseng, 2005).无机砷的生物可利用性, 环境毒性及在水体中的可移动性取决于它的存在形态.水中的无机砷主要以iAsⅤ(五价砷)和iAsⅢ(三价砷)形式存在(Liu et al., 2010), 这两种形态的无机砷在水体中的迁移、转化等环境行为受到诸多因素的影响, 其中天然有机质(Natural Organic Matter, NOM)就是控制无机砷迁移和转化过程的主要因素之一(Kalbitz et al., 1998; Harvey et al., 2002; Mcarthur et al., 2004; Wang et al., 2006; Bauer et al., 2006).
NOM是一种复杂且非均质的混合物, 包括非腐殖酸溶质如氨基酸、碳氢化合物、碳水化合物、脂肪、蜡、树脂、低分子量的酸和腐殖质(Tsuda et al., 2010), 广泛存在于水生环境(湖泊、河流、海洋、土壤水和地下水)、土壤环境中(Schumache et al., 2006; Hong et al., 2010).水体中NOM影响污染物砷的环境行为有几种方式.首先微生物降解的NOM可以还原被铁氧化物/氢氧化物包裹的砷, 使其溶解(McArthur et al, 2006).其次, 大量研究表明NOM分子结构中具有羟基、羧基、羰基等活性官能团, 很容易作为配位体与砷络合而改变砷的环境行为(Redman et al., 2002; Bauer et al., 2006).此外, 在其它重金属存在的情况下还可以通过架桥作用影响三者之间的络合体系从而改变砷的迁移行为(Mehmood et al., 2009; Sharma et al., 2010).腐殖质是NOM的重要组成部分, 在水环境的可溶性有机物中占比可达50%~95%(Adriano, 2001), 而胡敏酸(HA)和富里酸(FA)作为水体中腐殖质的两种主要类型, 因其吸附性和络合性等特征, 可以通过离子交换、表面吸附、配位络合等各类反应与金属和类金属发生作用(Redman et al., 2002; Lin et al., 2004).因此, 研究无机砷与这两种典型NOM的络合行为, 对于了解无机砷在自然水体中的迁移、转化等相关环境行为具有重要意义.
迄今为止, 关于无机砷与NOM络合物形成的推测已经形成, 但系统研究不同价态无机砷与不同类型NOM之间络合行为的研究相对较少(KoI et al., 2007).本文使用平衡透析实验装置分离自由态和络合态无机砷, 运用电感耦合等离子体质谱联用测定不同状态无机砷的浓度, 研究pH为中性条件下, 不同浓度(1、10、40 mg·L-1)iAsⅤ和iAsⅢ与不同浓度(0、10、20 mg·L-1)HA和FA之间的络合平衡时间, 测定不同种类和浓度NOM条件下iAsⅤ和iAsⅢ与NOM的络合比率, 以及不同比率iAs/ DOC条件下的条件分配系数Dom值.以期揭示无机砷与水体中典型NOM的络合行为, 从而更好阐述无机砷在水中迁移转化过程, 生物有效性等环境地球化学行为, 为更全面地评估砷的环境风险提供更有价值的数据和信息.
2 材料与方法(Materials and methods) 2.1 实验试剂与仪器所有实验溶液均用Milli-Q水进行制备(Millipore, 美国).实验中所用药品:HA(sigma-aldrich, 美国);FA(International Laboratory, 美国);iAsⅤ、iAsⅢ标准溶液(o2si smart solutions, 美国);HNO3(BV-Ⅲ级, 北京化学试剂研究所, 中国), 缓冲液所用药品购于sigma公司.实验中所用装置及仪器:透析袋(2000D, MWCO, 美国);PP烧杯(Nalgene, 美国);磁力搅拌器(雷磁, 中国);TOC分析仪(vario TOC select, elementar, 德国)电感耦合等离子体质谱(ICP-MS)(NexION300, PekinElmer, 美国).
2.2 NOM储液的制备取HA/FA约0.1 g溶于体积为50 mL的0.1 mol·L-1的NaOH溶液中, 避光振荡24 h, 使其完全溶解.调pH, 再用460 ℃灼烧后孔径为0.45 μm的GF/F玻璃纤维微孔滤膜(Whatman, 英国)进行过滤以制备HA/FA储备母液, 密封置于4 ℃冰箱中避光储存待用, 并取一定量HA/FA母液进行稀释, 采用TOC分析仪测定DOC浓度.
2.3 实验装置为了分离络合态无机砷与自由态无机砷, 采用平衡透析装置进行实验(装置示意如图 1所示).将透析袋一端折叠用夹子B夹紧使其不漏液, 另一端用夹子A固定在装满缓冲液的烧杯顶部.透析袋内加入NOM溶液(空白组不加NOM溶液).因NOM分子量大于透析袋的孔径, 即NOM分子及络合在NOM上的无机砷不能穿过透析袋.而自由态iAsⅢ和iAsⅤ则可以通过透析袋.实验开始后, 透析袋外部的自由态无机砷因袋内外浓度差扩散穿过透析袋, 与袋内NOM发生络合, 络合态无机砷则不能穿过透析袋释放到外部, 无机砷由袋外扩散到袋内的过程一直持续到络合完成, 且透析袋内外自由态无机砷浓度一致时结束.通过计算透析袋内外两侧之差即可算出络合态无机砷的比例以及无机砷与NOM络合的条件分配系数Dom.
 |
| 图 1 试验装置示意图 Fig. 1 The diagram of Experimental setup |
配制NOM母液2 g·L-1于NaOH中, 振荡过夜并在每次使用前超声30 min.分别配制浓度为0、10、20 mg·L-1的HA和FA并将pH调节至7.5±0.1, 使用针孔过滤器对溶液进行过滤(0.45 μm滤膜).过滤后溶液倒入透析袋内, 保证透析袋在缓冲液中保持垂直, 袋内保留少量空气, 使透析袋能悬浮于缓冲液中.透析袋外塑料烧杯缓冲液中加入iAsⅤ和iAsⅢ标准溶液适量, 配成浓度为1、10、40 mg·L-1的溶液.调节透析袋在塑料烧杯中位置, 使内外液面保持在同一水平面, 并避免透析袋贴于烧杯壁.用封口膜封住烧杯以防止实验过程中溶液蒸发损失, 保持烧杯中磁力转子转动进行搅拌混匀.实验开始后于1、2、4、12、24、48、72 h分别取透析袋内、外液1 mL, 利用电感耦合等离子体质谱仪对总砷浓度进行测定, 根据内外浓度差的变化情况得到络合平衡时间, 并测定络合态无机砷比例及Dom值.
2.5 数据分析与处理条件分配系数Dom(无机砷与NOM-水之间的分配系数, L·g-1)表征无机砷与NOM络合作用的强弱, 见式(1).
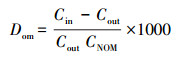
|
(1) |
式中, Cin为透析管内无机砷的浓度(mg·L-1), Cout为透析管外无机砷的浓度(mg·L-1), CNOM为实验中所使用NOM的浓度(mg·L-1).
采用SPSS 27.0统计学软件中one-way和two-way ANOVA两两比较方法(Tukey或Tamhane)分析数据显著性差异(当p < 0.05时接受).
3 结果与讨论(Results and discussion) 3.1 无机砷和NOM的络合平衡时间iAsⅤ与HA络合过程中Cin和Cout随时间变化如图 2所示.由图 2a~2c可以看出, 当HA浓度为0时, 透析袋外的iAsⅤ穿过透析袋膜, Cout随时间降低, 同时Cin随时间升高.0~12 h阶段中, Cout快速降低, 12 h时3个iAsⅤ处理组(C[iAs]Ⅴ=1、10和40 mg·L-1)透析袋内外无机砷浓度差已缩小至0.35、3.38和15.00 mg·L-1.12 h后Cin上升速度和Cout下降速度趋缓, 在48 h左右透析袋内外砷浓度基本达到一致.48~72 h之间, 随着时间的变化平衡状态保持不变.此时, 这3个iAsⅤ处理组透析袋内部无机砷浓度Cin分别为0.99、1.01和40.98 mg·L-1, 与Cout接近一致(1.01、1.02和40.44 mg·L-1).说明透析袋膜对溶液中iAsⅤ的移动性影响较小, 对系统的干扰极小.而从iAsⅤ与HA的络合过程来看, 72 h后透析袋内外浓度基本维持不变, 即已达到平衡状态.由此可见, 无HA情况下, iAsⅤ在系统内平衡时间大约为72 h.当在透析袋内添加10 mg·L-1及20 mg·L-1的HA时(2d~2i), Cin与Cout变化趋势与无HA处理组相似, 也分为快速扩散(0~12 h)、慢速扩散(12~48 h)和平衡(48~72 h)3个阶段, 72 h后保持浓度不变.但与HA空白处理组不同的是, 当Cin和Cout在48 h后趋于平衡时, Cin高于Cout.这一方面验证了透析袋内的iAsⅤ与HA发生了络合作用, 另一方面说明透析袋内外浓度平衡基本在72 h内完成, 与不添加HA平衡时间一致.
 |
| 图 2 iAsⅤ与HA络合过程(a、d、g为浓度1 mg·L-1的iAsⅤ, b、e、h为浓度10 mg·L-1的iAsⅤ, c、f、1为浓度40 mg·L-1的iAsⅤ;a、b、c不添加HA, d、e、f中HA浓度为10 mg·L-1, g、h、i中HA浓度为20 mg·L-1) Fig. 2 The complexation Process of arsenate iAsⅤ with HA |
iAsⅤ与FA络合过程呈现出与HA络合类似的趋势.以FA浓度10 mg·L-1, iAsⅤ浓度为10 mg·L-1(C[FA]=10 mg·L-1, C[iAs]Ⅴ=10 mg·L-1)和FA浓度20 mg·L-1, iAsⅤ浓度为40 mg·L-1(C[FA]=20 mg·L-1, C[iAs]Ⅴ=40 mg·L-1)为例(3a、3b).Cin和Cout在12 h后变化趋缓, 48 h后浓度维持不变, 可以认为72 h达到平衡, 此时C[FA]为10 mg·L-1, C[iAs]Ⅴ为10 mg·L-1处理组中Cin和Cout分别为10.50 mg·L-1和9.92 mg·L-1;C[FA]为20 mg·L-1, C[iAs]Ⅴ为40 mg·L-1处理组中Cin和Cout分别为43.01 mg·L-1和40.84 mg·L-1.
 |
| 图 3 iAsⅤ与FA络合过程(a为FA浓度10 mg·L-1、iAsⅤ浓度10 mg·L-1, b为FA浓度20 mg·L-1、iAsⅤ浓度40 mg·L-1) Fig. 3 The complexation Process of arsenate iAsⅤ with FA |
iAsⅢ与两种NOM络合过程中Cin和Cout随时间变化如图 4所示.不添加NOM处理组以C[iAs]Ⅲ为10 mg·L-1和40 mg·L-1为例(图 4a、4b), 1 h时后Cin已达5.32 mg·L-1和13.89 mg·L-1, 而对于相同浓度的iAsⅤ处理组, 同样时间后Cin只有0.57 mg·L-1和2.08 mg·L-1, 也就是说相同浓度的iAsⅢ的扩散速度要高于iAsⅤ.iAsⅢ处理组的快速变化阶段在4 h后结束, 4 h后C[iAs]Ⅲ为10 mg·L-1和40 mg·L-1处理组中渗透膜内外浓度差已缩小至0.03 mg·L-1和4.14 mg·L-1.4 ~24 h进入慢速变化阶段, 24 h后Cin和Cout浓度基本维持不变, 48 h即可达到平衡状态, 这也较iAsⅤ处理组时间更短, 内外平衡浓度约分别为1.00 mg·L-1和40.57 mg·L-1.添加HA和FA与iAsⅢ络合同样以C[iAs]Ⅲ为10 mg·L-1和40 mg·L-1为例(4c和4d, 4e和4f), Cin和Cout呈现出的变化趋势与不添加NOM处理组变化情况相似, 0 ~4 h中Cin快速随时间增长, 4~24 h中Cin增长速度趋缓, 48 h基本可以达到平衡状态.与不添加NOM处理组有区别的是, 添加HA和FA处理组在两个iAsⅢ浓度下(10 mg·L-1和40 mg·L-1), 24 h时都出现了Cin高于Cout的现象, 这也验证了HA和FA与iAsⅢ发生了络合, 并在48 h后基本达到平衡状态.因此, 可以确定两种NOM与iAsⅢ的络合时间为48 h.
 |
| 图 4 iAsⅢ与HA/FA络合过程(a和b中不添加NOM, c和d中HA浓度分别10 mg·L-1和20 mg·L-1, e和f中FA浓度分别为10 mg·L-1和20 mg·L-1;a、c、e中iAsⅢ浓度为10 mg·L-1, b、d、f中iAsⅢ浓度为40 mg·L-1) Fig. 4 The complexation Process of arsenite iAsⅢ with HA/FA |
大量文献认为水中的无机砷可以与NOM发生络合反应(Harvey et al., 2002;Bauer et al., 2006; Sharma et al., 2010), 而无机砷更是可以与溶解有机质(DOM)直接连接形成二元络合物(As-DOM), 这与DOM的多种有机官能团, 例如酚基、羧基、羟基、巯基和氨基的存在有关(Redman et al, 2002; Lin et al, 2004).无机砷与NOM的络合时间与砷形态及环境条件有关, 现有研究一般认为无机砷与NOM络合作用可以在数小时到数天内达到平衡(Warwick et al, 2005;Liu et al, 2010; Liu et al, 2011).一般情况下, iAsⅢ与NOM的络合作用要快一些(Liu et al, 2010).Warwick等认为无机砷与HA的络合可在24 h内达到平衡(Warwick et al, 2005).与此相似, Liu等观察到iAsⅢ与HA的络合在20 h左右可达到平衡(Liu et al, 2010).此外, 鄢元波等采用同样实验方法得到与砷同主族的重金属锑络合HA时间约为72 h(鄢元波等, 2013).这些结果与本研究得到的平衡时间都较为接近.
3.2 两种无机砷与HA的络合作用通过平衡状态时络合态砷占总砷的比例可以反映两种无机砷与NOM的络合情况.当HA浓度为10 mg·L-1时(图 5a), 与HA络合的iAsⅢ和iAsⅤ比例范围分别为4.09%~5.79%和5.51%~11.14%.在HA浓度增大到20 mg·L-1时(图 5b), 络合态无机砷比例相应增大(p < 0.05).此时与HA络合的iAsⅢ和iAsⅤ比例范围分别为4.40%~6.32%和7.71%~12.25%.Hoda等在无机砷与HA络合实验中也得到了相同的结论(Hoda et al., 2014), 两种无机砷iAsⅢ和iAsⅤ都可以与HA发生络合反应, 并且络合态砷的比例随HA浓度升高而增加, 文章根据结合位点模型推测结合位点的数目与HA的浓度成正相关.目前的研究认为无机砷与金属类似, 可以直接与HA的官能团发生络合作用(Watanabe et al., 1994; Bauer et al., 2009).对于iAsⅢ, 当pH < 9时, 通常以不带电荷的形式如As(OH)3出现, 可与HA中的酚基发生配体交换反应(Buschmann et al., 2015).而对于iAsⅤ, pH为中性时常以H2AsO4-和HAsO42-阴离子基团的形式存在, 由于带负电荷, 其在水体中通过与HA胺基官能团发生弱绑定作用而发生络合(Tipping, 2002).
 |
| 图 5 络合态与自由态砷占总砷比例(a图中HA浓度为10 mg·L-1, b图中HA浓度为20 mg·L-1, c图中FA浓度为10 mg·L-1, d图中FA浓度为20 mg·L-1) Fig. 5 Proportion of the bounded arsenic and free arsenic to total |
从图 5a和5b中还可以看出, 浓度相同情况下, 相应iAsⅤ浓度处理组与HA络合态比例都要高于同样浓度iAsⅢ处理组(p < 0.05).以C[HA]为10 mg·L-1为例, 当C[iAs]Ⅴ与C[iAs]Ⅲ同为1 mg·L-1时, 与HA络合比例分别为11.14%与5.79%.这样的结论与Buschmann等和Warwick等的研究报道一致(Warwick et al., 2005; Buschmann et al., 2015).研究认为iAsⅤ与iAsⅢ在与HA络合过程中存在着不同的络合机制是产生差异的原因(Buschmann et al., 2015).虽然iAsⅤ以阴离子基团存在, 与HA络合应属于弱绑定.但也有理论推测, 可能是由于iAsⅤ中心较高的电荷以及其他形式的螯合和稳定作用造成络合加强(Watanabe et al., 1994).这一理论认为当配位体的个数小于6个时, 可能在金属带正电荷中心发生关联配体交换机制(Park et al., 2006).因为iAsⅤ中心存在着+V价的形式电荷, 就会在其亲电中心位点上加入酚基, 随后发生质子化作用进而产生释水.虽然两种化合物整体都是带有负电荷的, 但由于酚供体特性, 以及与其它官能团产生的螯合作用以及氢键等作用, iAsⅤ和HA的结合会更加稳定.此外, Warwick等的研究报道中认为iAsⅤ在络合过程中形成的是双层结构, 而iAsⅢ的络合物中则没有出现这样的结构(Warwick et al., 2005).
3.3 两种无机砷与FA的络合作用FA是除了HA外水体中另一种常见的NOM, 通过同样的方法得到平衡状态时络合态砷占总砷的比例见图 5c和5d.与HA相似, 两种无机砷与FA的络合比例同样随FA浓度提高而增加(p < 0.05).当FA浓度为10 mg·L-1时(图 5c), 与FA络合的iAsⅢ和iAsⅤ比例范围分别为3.06%~5.02%和4.46%~8.42%.在FA浓度增大到20 mg·L-1时(图 5d), 比例范围分别提高到3.65%~5.81%和5.18%~10.41%.Hoda等在对无机砷与NOM的络合研究中认为, 无机砷与FA络合过程同其与HA络合模式相似, 都为强弱两类结合位点结合模型, 而这两类结合位点的数目都与NOM的浓度呈正相关(Hoda et al., 2014).
此外, 将FA与两种无机砷的络合情况与HA相比可以发现, 相应浓度HA与各无机砷处理组的络合比例都要高于同样浓度FA与同种无机砷处理组的络合比例(p < 0.05).以C[iAs]Ⅲ和C[iAs]Ⅴ同为40 mg·L-1为例, C[HA]为10 mg·L-1浓度下iAsⅢ和iAsⅤ与其络合比例为4.09%和5.51%, 而同浓度情况下, iAsⅢ和iAsⅤ与FA的络合比例只有3.06%和4.46%.换言之, FA与两种无机砷的络合能力要低于HA.我们认为这样的现象可能与HA和FA的结构有关.HA与FA都是复杂的有机酸混合物, 从分子量上看, FA的分子量小于HA, 其C、N含量低于HA, 而O、S的含量高于HA, 氧化程度高于HA.从分子结构上看, FA的缩合度低于HA, 分子结构没有HA复杂.正因为较高的分子量和较复杂的分子结构使得HA比FA可能拥有更多的结合位点和更强的结合能力(Tan, 2003; Ting et al., 2016).
从两种NOM与无机砷的络合行为可以看出, 在C[NOM]较高的水体中, 较多的无机砷会与NOM发生络合, 这会导致iAsⅤ和iAsⅢ在水体中的迁移能力改变, 可能在一定程度上可降低其生物可利用性.这也意味着开发利用高C[NOM]的生物吸附剂或污泥来处理低浓度的含砷废水具有一定的应用前景.
3.4 不同比率iAs/ DOC对无机砷与HA/FA络合作用的影响图 6给出了不同iAs/ DOC比率下, iAsⅤ和iAsⅢ与两种NOM的络合条件分配系数Dom情况.对于iAsⅤ, 由图 6a和6b可见, 不论与HA或FA络合, Dom都随着iAsⅤ/ DOC比值的上升而下降.与HA络合过程中Dom值从13.05 L·g-1下降到5.67 L·g-1, 与FA络合的Dom值则从10.98 L·g-1下降到4.56 L·g-1.这样下降的现象与单位iAsⅤ可以发生络合配位点数目减少有关(Buschmann et al., 2015).此外, 可以从图中看出, Dom值的下降不能通过简单的线性曲线进行拟合, 整个过程呈现出至少包含两个不同斜率的线性方程.将两阶段分别进行线性拟合, 得到HA的斜率分别为-25.20和-1.43, FA的斜率值则分别为-43.77和-0.72.也就是呈现出先快速下降后缓慢下降两个下降速率有较大差异的阶段.这样的现象说明在实验所进行的不同iAsⅤ/ DOC比率范围内, 至少存在着两种不同类型的络合点类型.同样的情况也出现在iAsⅢ与HA/FA络合中(图 6c和6d).iAsⅢ与HA络合过程中随iAsⅢ/ DOC比率上升Dom值从6.53 L·g-1下降到4.18 L·g-1, 与FA络合的Dom值则从5.99 L·g-1下降到3.11 L·g-1.HA拟合两阶段线性比率分别为-11.19和-0.36.FA中的比率则是-16.79和-0.42.这种在无机砷与NOM络合过程中呈现出的随无机砷浓度(或是iAs/DOC比率)非线性下降的结论也在文献中有过报道(Hoda et al., 2014; Buschmann et al., 2015).这可能与NOM结构复杂有关, NOM含有多种官能团, 而这些官能团具有不同络合砷的能力(刘广良等, 2011).Buschmann等的文章中认为在无机砷与NOM络合中存在不同的络合类型(Buschmann et al., 2015).而Hoda等则用双位配体模型拟合无机砷与NOM的络合数据, 得到了较好的结果(Hoda et al., 2014).因此推测无机砷与NOM的络合过程中可能存在两种类型的络合点位, 其中一种络合能力较强, 可以形成较为稳定的络合, 而另一种络合能力较弱, 形成的络合物稳定性不如前者.
 |
| 图 6 不同比率iAs/DOC下无机砷与NOM条件分配系数(a、b为iAsⅤ, c、d为iAsⅢ, a、c为HA, b、d为FA) Fig. 6 Dom of arsenic to HA/FA under different iAs/DOC ratios |
1) iAsⅤ与两种NOM(HA/FA)络合在72 h可达到络合平衡状态, 而由于iAsⅢ具有较iAsⅤ更快的扩散速度, 48 h即可达到络合平衡.
2) HA因为更高的分子量和更加复杂的分子结构, 更易与两种无机砷络合, 但不论是HA还是FA, 与无机砷络合作用强度都随NOM浓度的增高而增加.
3) 无机砷与HA/FA络合分配常数随iAs/ DOC比率的上升而呈现出非线性的下降趋势, 这样非线性的现象可能由于至少两类络合位点的类型有关.
4) 无机砷与NOM的络合是一种不可忽视的影响砷环境行为的过程.环境中有机质的存在可能会影响砷的存在形态继而影响砷的移动性和生物可利用性.
Adriano D C. 2001. Trace elements in terrestrial environments:Biogeochemistry, bioavailability and risks of metals(2nd ed)[M]. New York: Springer.
|
Anawar H M, Akai J, Komaki K, et al. 2003. Geochemical occurrence of arsenic in groundwater of Bangladesh:sources and mobilization processes[J]. Journal of Geochemical Exploration, 77(2): 109–131.
|
Bauer M, Blodau C. 2006. Mobilization of arsenic by dissolved organic matter from iron oxides, soils and sediments[J]. Science of the Total Environment, 354(2): 179–190.
|
Bauer M, Blodau C. 2009. Arsenic distribution in the dissolved, colloidal and particulate size fraction of experimental solutions rich in dissolved organic matter and ferric iron[J]. Geochimica et Cosmochimica Acta, 73: 529–542.
DOI:10.1016/j.gca.2008.10.030
|
Buschmann J, Kappeler A, Lindauer U, et al. 2015. Arsenite and arsenate binding to dissolved humic acids:influence of pH, type of humic acid, and aluminum[J]. Environmental Science and Technology, 40(19): 6015–6020.
|
Harvey C F, Swartz C H, Badruzzaman A B, et al. 2002. Arsenic mobility and groundwater extraction in Bangladesh[J]. Science, 298(5598): 1602–1606.
DOI:10.1126/science.1076978
|
Hoda F, Lin T F. 2014. Experimental determination and modeling of arsenic complexation with humic and fulvic acids[J]. Journal of Hazardous Materials, 279: 569–578.
DOI:10.1016/j.jhazmat.2014.07.039
|
Hong S W, Kim H S, Chung T H. 2010. Alteration of sediment organic matter in sediment microbial fuel cells[J]. Environmental Pollution, 158(1): 185–191.
DOI:10.1016/j.envpol.2009.07.022
|
Kalbitz K, Wennrich R. 1998. Mobilization of heavy metals and arsenic in polluted wetland soils and its dependence on dissolved organic matter[J]. Science of the Total Environment, 209(1): 27–39.
DOI:10.1016/S0048-9697(97)00302-1
|
KoI D A P, Kim J Y. 2007. Effect of contact order on the adsorption of inorganic arsenic species onto hematite in the presence of Humic acid[J]. Journal of Hazardous Materials, 141: 53–60.
DOI:10.1016/j.jhazmat.2006.06.084
|
Lin H T, Wang M C, Li G C. 2004. Complexation of arsenate with humic substance in water extract of compost[J]. Chemosphere, 56(1): 1105–1112.
|
Liu C P, Luo C L, Gao Y, et al. 2010. Arsenic contamination and potential health risk implications at an abandoned tungsten mine, southern China[J]. Environmental Pollution, 158(3): 820–826.
DOI:10.1016/j.envpol.2009.09.029
|
Liu G, Cai Y. 2010. Complexation of arsenite with dissolved organic matter:conditional distribution coefficients and apparent stability constants[J]. Chemosphere, 81: 890–896.
DOI:10.1016/j.chemosphere.2010.08.002
|
Liu G, Fernandez A, Cai Y. 2011. Complexation of arsenite with humic acid in the presence of Ferric Iron[J]. Environmental Science and Technology, 45(8): 3210–3216.
DOI:10.1021/es102931p
|
刘广良, 蔡勇. 2011. 环境中砷与溶解有机质的络合作用[J]. 环境化学, 2011, 30(1): 50–55.
|
Mandal B K, Suzuki K T. 2002. Arsenic round the world:a review[J]. Talanta, 58(1): 201–235.
DOI:10.1016/S0039-9140(02)00268-0
|
Matschullat J. 2000. Arsenic in the geosphere-a review[J]. Science of the Total Environment, 249(1): 297–312.
|
McArthur J M, Ravenscroft P, Safiulla S, et al. 2001. Arsenic in groundwater:Testing pollution mechanisms for sedimentary aquifers in Bangladesh[J]. Water Resource Research, 37: 109–117.
DOI:10.1029/2000WR900270
|
Mcarthur J M, Banerjee D M, Hudson-Edwards K A, et al. 2004. Natural organic matter in sedimentary basins and its relation to arsenic in anoxic ground water:the example of West Bengal and its worldwide implications[J]. Applied Geochemistry, 19(8): 1255–1293.
DOI:10.1016/j.apgeochem.2004.02.001
|
Mehmood A, Hayat R, Wasim M, et al. 2009. Mechanisms of arsenic adsorption in calcareous soils[J]. International Journal of Agriculture and biology, 1: 59–65.
|
Naidu R, Smith E, Owens G, et al. 2006. Managing Arsenic in the Environment:From Soil to in the Environment:From Soil to Human Health[M]. Melbourne: CSIRO publishing.
|
Park J K, Kim B G. 2006. Potential energy surfaces for ligand exchange reactions of square planar diamagnetic pty2l2 complexes:hydrogen bond versus apical interaction[J]. Bulletin of the Korean Chemical Society, 27: 1405–1417.
DOI:10.5012/bkcs.2006.27.9.1405
|
Redman A D, Macalady D L, Ahmann D. 2002. Natural organic matter affects arsenic speciation and sorption onto hematite[J]. Environmental Science and Technology, 36: 2889–2896.
DOI:10.1021/es0112801
|
Schumacher M, Christl I, Vogt R D, et al. 2006. Chemical composition of aquatic dissolved organic matter in five boreal forest catchments sampled in spring and fall seasons[J]. Biogeochemistry, 80(3): 263–275.
DOI:10.1007/s10533-006-9022-x
|
Sharma P, Ofner J, Kappler A. 2010. Formation of binary and ternary colloids and dissolved complexes of organic matter, Fe and As[J]. Environmental Science and Technology, 44: 4479–4485.
DOI:10.1021/es100066s
|
Smedley P L, Kinniburgh D G. 2002. A review of the source, behaviour and distribution of arsenic in natural waters[J]. Applied Geochemistry, 17(5): 517–568.
DOI:10.1016/S0883-2927(02)00018-5
|
Tan K H. 2003. Humic matter in soil and the environment, in:principles and contro-versies[M]. New York: CRC Press.
|
Ting C, Chen A, Zeng Y H, et al. 2016. Association between arsenic and different-sized dissolved organic matter in the groundwater of black-foot disease area, Taiwan[J]. Chemosphere, 159: 214–220.
DOI:10.1016/j.chemosphere.2016.06.007
|
Tipping E. 2002. Cation binding by humic substances[M]. Cambridge: Cambridge University Press: pp. 12.
|
Tseng C H. 2005. Blackfoot disease and arsenic:a never-ending story[J]. Journal of Environmental Science & Health Part C, 23(1): 55–74.
|
Tsuda K, Mori H, Asakawa D, et al. 2010. Characterization and grouping of aquatic fulvic acids isolated from clear-water rivers and lakes in Japan[J]. Water Research, 44(13): 3837–3846.
DOI:10.1016/j.watres.2010.04.038
|
Wang S, Mulligan C N. 2006. Effect of natural organic matter on arsenic release from soils and sediments into groundwater[J]. Environmental Geochemistry and Health, 28(3): 197–214.
DOI:10.1007/s10653-005-9032-y
|
Warwick P, Inam E, Evans N. 2005. Arsenic's interaction with humic acid[J]. Environmental Chemistry, 2: 119–124.
DOI:10.1071/EN05025
|
Watanabe A, Itoh K, Arai S, et al. 1994. Comparison of the composition of humic and fulvic acids prepared by the IHSS method and NAGOYA method[J]. Soil Science and Plant Nutrition, 40: 601–608.
DOI:10.1080/00380768.1994.10414299
|
鄢元波, 孙福红, 吴丰昌, 等. 2013. Sb(Ⅲ)与腐殖酸络合特征[J]. 环境科学研究, 2013, 26(3): 307–312.
|