 2019, Vol. 39
2019, Vol. 39



2. Laboratory of Environmental Technology, Institute of Materials and Environment Technology, Tallinn University of Technology, Tallinn 19086
2. Laboratory of Environmental Technology, Institute of Materials and Environment Technology, Tallinn University of Technology, Tallinn 19086
厌氧氨氧化工艺将是未来主导的废水脱氮工艺(Pan et al., 2019), 但厌氧氨氧化菌追求的水质条件为C/N比小于2.0, NO2-/NH3大于1.32, 废水中的有机氮需要彻底氨化.在焦化废水和金矿提炼废水中, 经常出现高浓度的硫氰酸盐和氰化物, 微生物对微量氰化物的解毒作用可以将其转化为硫氰酸盐(Ramirez et al., 2002; Gupta et al., 2010).硫氰酸盐作为一种具有中等毒性且化学性质稳定的污染物, 在焦化废水中的浓度普遍处于200~1000 mg·L-1范围内, 是含量最高的无机污染物, 也是构成TN的主要含氮化合物(Boucabeille et al., 1994a; 1994b; 韦朝海, 2012).基于很强的铁络合能力, 通过化学沉淀、絮凝、吸附和厌氧消化等手段无法从废水中有效去除硫氰酸盐(Gould et al., 2012).目前, 主要采取生物方法和化学氧化法对硫氰酸盐的浓度加以控制.生物法利用有限的一些微生物以不同的代谢方式降解硫氰酸盐, 如以硫氰酸盐作为电子供体的自养反硝化途径, 或以硫氰酸盐作为硫与氮来源的异养途径(Pan et al., 2018; Sun et al., 2019).硫氰酸盐具有还原性, 使用ClO-、O3、H2O2、FeO42-、Fenton试剂等氧化剂可以实现其转化, 但难以彻底去除(Young, 2001; Van Leeuwen et al., 2003; Sharma et al., 2008).尽管这些技术可以转化废水中的硫氰酸盐, 但也存在一些弊端:与Fe3+和Cu2+等金属离子的络合物为难降解的显色物质;过量消耗化学药剂, 反应不彻底, 导致处理费用高;不能完全去除TN.
脉冲电晕放电技术(PCD)(Malik et al., 2001)是一种新型的高级氧化技术, 放电时间较短, 总持续时间仅有100 ns.以脉冲的形式在不均匀排列的电极之间进行不击穿电流的电晕放电, 作用于流向电极之间的废水(Pokryyailo et al., 2006; Gerrity et al., 2010; Dobrin et al., 2013).放电过程可以形成一些高能电子、离子和激发态粒子等低温等离子通道, 存在多种物理场与氧化剂之间的协同作用.原位产生的O3及·OH等多种活性氧化物种, 具有很强的氧化活性.所产生的活性氧化物种的反应方程式(Liu et al., 2018)如下:

|
(1) |
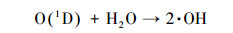
|
(2) |

|
(3) |
本研究尝试使用PCD技术, 以配制的SCN--N模拟废水作为试验对象, 分析PCD放电处理过程中SCN--N、CN--N、NO3--N、NH4+-N、NO2--N、SO42-和TOC的浓度变化, 基于动力学常数的差异分析pH、SO42-和HCO3-对NH4+-N降解的影响, 明确SCN--N分解过程中氮的动态转化和影响因素, 有利于获得目标的水质.
2 材料与方法(Materials and methods) 2.1 实验装置采用课题组自主搭建的脉冲电晕放电(PCD)小试装置进行实验, 装置与实验工艺流程如图 1所示.该装置由主体反应区、高压脉冲发生器及带有循环泵的蓄水池组成.脉冲电晕反应区所使用的电极为线-板式电极, 其中高压电极的总长度为12 m, 包含10个高压线状电极, 平行排列, 高压电极电极间距为30 mm, 高压电极和接地电极板之间的距离为20 mm;反应器还包括变频循环水泵、空气压缩机(用于反应区通风及调节反应区气体组成比例)、高压分压器等.该放电装置的脉冲持续时间为100 ns, 输入功率220 V, 峰值电压18 kV, 峰值电流为380~400 A;可调节的脉冲重复频率范围是100~1000 pps(pulse per second).每个脉冲输入能量为0.22 J, 实验中使用的脉冲放电频率分别为200和800脉冲/秒(pps), 以功率计的脉冲宽度为22~220 W.所处理溶液通过循环泵提升到达反应器顶部, 然后自由喷洒而下, 反应器顶部有尺寸为420 mm×40 mm的分布板且布有230个直径为1.0 mm的小孔, 溶液通过小孔形成小液滴水雾, 直流向下穿过放电反应区, 循环流速稳定在1.0 m3·h-1.
 |
| 图 1 反应装置与工艺示意图 Fig. 1 Pulsed corona discharge experimental device |
为考察经PCD处理过程中污染物随时间的动态转化规律, 以及硫氰酸盐分解副产物(SO42-和HCO3-)对氨氮转化的影响, 在脉冲重复频率为800 pps的条件下开展了动力学实验, 因为800 pps较200 pps反应速率更快, 同样的能量输出下, 200 pps条件下所需的放电处理时间是800 pps条件下的4倍.对此, 实验过程按照如下步骤操作:
① 在不同pH(pH=4、7、10)条件下, 对硫氰酸钾配制的模拟废水进行试验, 模拟废水中SCN--N浓度为25 mg·L-1, 通过SCN--N、CN--N、NO3--N、NH4+-N、NO2--N、SO42-和TOC的测定分析在放电处理过程中氮存在形态的变化;
② 在不同pH(pH=6、8、9、10、11)条件下, 对氯化铵配制的模拟废水进行试验, 模拟废水中NH4+-N浓度为25 mg·L-1, 通过NO3--N、NH4+-N、NO2--N的测定分析在放电过程中NH4+-N的转化, 并进行降解动力学拟合.
③ 采用初始NH4+-N浓度为25 mg·L-1, 且pH控制为10.0的条件下, 评估不同浓度SO42-和HCO3-对NH4+-N分解动力学的影响.
实验开始时, 在储水罐(图 1)中配置好所需浓度的模拟废水, 体积为20 L, 开启循环泵, 循环3 min, 使水样充分均匀, 取样(未处理的初始水样), 然后开始放电试验, 在取样时间点停止放电后取样, 以此类推, 直至实验结束.实验过程中, 循环泵一直处于开启状态, 废水处于不断循环的状态.实验过程中使用1.0 mol·L-1的HCl或NaOH溶液调节pH值.每间隔一定时间从取样口收集50 mL水样, 进行水质指标的测定.
2.3 分析方法溶液pH值采用三星SX731型便携式pH计(中国)进行测定;总有机碳(TOC)使用TOC分析仪(TOC-Vcpn, Shimadzu, 日本)定量分析;NO3-、NO2-和SO42-等无机阴离子使用离子色谱(ICS-900, Dionex, 美国)测定;SCN-采用铁(Ⅲ)-硫氰化物分光光度法、CN-采用吡啶-巴比妥酸分光光度法、NH4+-N采用纳氏试剂分光光度法测定.
2.4 能效计算采用式(4)计算污染物在不同条件下分解的能效(Ajo et al., 2015; Liu et al., 2018):
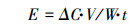
|
式中, △C指污染物浓度的变化(g·m-3);V指模拟废水体积(m3);W指反应器放电输入功率(kW);t指放电处理时间(h).
3 结果与讨论(Results and discussion) 3.1 SCN-的PCD氧化过程中pH值的影响pH值对PCD运行过程中产生活性物质的种类和速率具有显著影响, 且臭氧在水相中的分解速率也取决于溶液的pH(Hoigné et al., 1983;Meunier et al., 2006).臭氧的分解主要依靠与溶液中OH-反应, 如反应式(5)及反应式(6)所示.
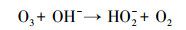
|
(5) |
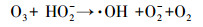
|
(6) |
因此, 为了评估pH值对PCD分解硫氰化物的影响, 以及溶液中氮存在形式随时间的变化, 配制了25.00 mg·L-1 SCN--N的模拟废水, 在pH为4.0、7.0和10.0时放电处理180 min.如图 2所示, 在3个不同pH的反应体系中, SCN--N的浓度均迅速降低, 反应30 min后均未被检测出.伴随着SCN--N的分解, CN-浓度呈现出先增高后降低的趋势.这是因为当O3的优先反应物SCN--N被分解完全后, 会进一步把CN-氧化为CNO-.CNO-的化学状态极不稳定, 易被水解为NH4+-N和HCO3-, 或被O3进一步氧化为N2, 两类反应受到体系中O3量和酸碱度的双重影响(Mirat et al., 1985; Ko et al., 2009; McKay et al., 2011; Kornev et al, 2017).这也解释了放电反应20 min后, 体系中NH4+-N逐渐累积, 且pH=7.0及10.0条件下氮的总量有所减少的现象.此外, 在pH=4.0的条件下, 体系中氮全部以NH4+-N的形式存在, 而在pH=7.0及10.0的条件下, 最终产物为NH4+-N和NO3--N共存.这是由于酸性条件下PCD产生的活性物质主要以O3形式存在, 氨氮主要以NH4+-N形式存在, 而O3主要与未电离态的氨氮(NH3)反应.由以上结果, 结合TOC和SO42-浓度的变化趋势, 可以明确SCN--N在PCD技术作用下的分解途径, 可表示如下:
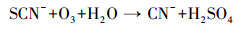
|
(7) |
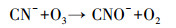
|
(8) |
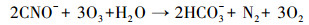
|
(9) |

|
(10) |
 |
| 图 2 SCN-的PCD处理中pH值的影响 Fig. 2 Treatment of wastewater containing SCN- by PCD under different pH conditions |
表 1列举了SCN--N的拟合分解动力学方程及其分解能效.当反应体系的pH为4、7和10时, SCN--N分解的反应速率常数分别为0.9545、1.094和1.226 min-1.明显地, SCN--N的分解速率随着pH的升高而增大.换言之, 碱性条件更有利于反应过程的进行, 这主要是由于碱性环境中部分O3会分解转化为·OH等活性物质, ·OH的氧化能力较高, 加速了SCN--N的氧化.值得注意的是, 3个反应体系中SCN--N的分解速率均远远高于文献报道的关于SCN--N的生物性降解速率(Pan et al., 2018).且当反应体系的pH为4、7和10时, SCN--N的分解能效分别为6.52、7.49、9.77 g·kW-1·h-1, 即pH越高, SCN-的分解能效越高, 分解速率越快, 这与速率常数分析所得结果相一致.
| 表 1 不同pH条件下拟合的SCN--N分解动力学方程及其分解能效 Table 1 SCN--N decomposition kinetic equations fitted and energy efficiency under different pH conditions |
NH4+-N被认为是还原性相对较弱的化合物, 目前已知仅好氧氨氧化菌和厌氧氨氧化菌可生物性氧化NH4+-N.化学法中, NH4+-N几乎不被重铬酸钾氧化, 仅O3和·OH等高活性物质才可利用.其中, 厌氧氨氧化被认为是最节能和高效的NH4+-N去除方式, 是未来水处理技术的核心方向.PCD对SCN--N废水有较高的氨解速率, 为厌氧氨氧化工艺的构建提供了必备的底物条件.此外, SCN--N分解过程中产生的HCO3-, 也可作为厌氧氨氧化菌代谢过程的碳源, 从而避免了外部无机碳源的投加.
3.2 pH对氨氮分解动力学的影响考虑到空气吹脱可能对溶液中NH4+-N浓度的影响, 在其他实验条件相同, 但不开启放电装置且pH为11的条件下, 进行了吹脱实验.如图 3所示, NH4+-N浓度随反应的进行而少许下降, 反应120 min后NH4+-N的最大降解量为1.8 mg·L-1, 相应的NH4+-N最大去除率为7.2%.由于pH的增大利于溶液中NH4+-N的吹脱, 故PCD处理NH4+-N废水过程中, 因氮吹脱导致的NH4+-N去除率应小于10%.
 |
| 图 3 空白实验中氨氮浓度及去除率的变化 Fig. 3 Concentration and removal rate of ammonia nitrogen in blank experiment |
从图 2中可知, 在酸性条件下NH4+-N并没有被氧化为NO3--N, 而中性及碱性条件下情况并不同, 由此可知pH对NH4+-N分解的影响较为显著, 有必要深入考察.为探究pH对NH4+-N分解的影响, 配制NH4+-N浓度为25.00 mg·L-1的模拟废水, 在pH为6.0、8.0、9.0、10.0和11.0时经PCD放电处理120 min, NH4+-N的分解情况如图 4所示, 表 2为不同pH条件下NH4+-N分解的拟合动力学方程及其分解能效.
 |
| 图 4 不同pH条件下PCD对NH4+-N的降解 Fig. 4 Degradation of NH4+-N by PCD under different pH conditions |
| 表 2 不同pH下NH4+-N降解的拟合动力学方程及其分解能效 Table 2 Fitting kinetic equations and energy efficiency for the degradation of NH4+-N under different pH conditions |
从图 4及表 2中速率常数及分解能效的计算可知, NH4+-N的分解符合动力学零级模型, 可决系数均在0.98以上, 表明模型符合实际氧化处理过程中NH4+-N浓度的变化曲线.pH值对NH4+-N的分解转化影响显著, 总体来说, O3分解NH4+-N的能力会随着pH的升高而变强.形成这个结果的原因是, 氨氮在水溶液中有游离的氨(NH3)与铵根离子(NH4+)两种存在形式, 两者可以互相转化.pH值对两种存在形式所占的比例有很大的影响, pH越高, NH3所占比重越大, 反之, pH较低时NH4+比重更高.当pH为7.0时, 氨氮主要以NH4+形式存在, 当pH升至11.0时, NH3的存在比值将超过90%(Khuntia et al., 2012).除此之外, O3与NH3的反应速率常数为20 L·mol-1·s-1, 而O3与NH4+的反应速率常数仅为1 L·mol-1·s-1 (Hoigné et al., 1985).pH值的增大, 也可以加速O3转化为氧化性更强的·OH等活性物质.
随着NH4+-N的分解, NO3--N的浓度逐渐升高, 结合动力学常数可知, NO3--N的生成速率常数与NH4+-N的降解速率常数基本相同, 且总氮浓度守恒.例如, 当pH为10.0时, NO3--N的生成速率常数为0.2068 min-1, 略低于NH4+-N的降解速率常数0.2134 min-1.这表明O3和·OH等活性物质可以氧化NH4+-N, 且NH4+-N几乎完全转化为NO3--N;此过程可能存在的中间产物NO2--N, 但NO2--N极易被氧化为NO3--N, 反应式如下:
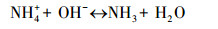
|
(11) |
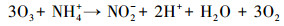
|
(12) |
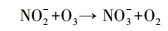
|
(13) |

|
(14) |
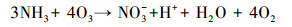
|
(15) |
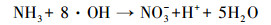
|
(16) |
由此可见, PCD能够氧化不能被重铬酸钾所氧化的NH4+-N(Bunce et al., 2011; Kim et al., 2006; Chen et al., 2007; Bejan et al., 2013; 韦聪等, 2018).同时, 可证实SCN--N在pH为7.0和10.0的氧化分解过程中, NO3--N是来自于O3对NH4+-N的氧化(Luo et al., 2015).此外, 同等pH条件下, SCN--N分解过程中NH4+-N的降解速率远远低于单独NH4+-N溶液中的降解速率, 可能的原因是·OH被某些抑制剂所影响.
3.3 SO42-及HCO3-浓度对NH4+-N分解动力学的影响为了验证SCN--N分解过程产生的SO42-和HCO3-在PCD工艺中对NH4+-N分解的影响, 实验选取NH4+-N初始浓度为25.00 mg·L-1、pH为10.0、不同SO42-和HCO3-的浓度条件下, 放电处理120 min, 实验结果如图 5所示, 表 3为不同的SO42-和HCO3-浓度下NH4+-N分解的拟合动力学方程及其分解能效.
 |
| 图 5 不同浓度的SO42-和HCO3-下PCD对NH4+-N的降解 Fig. 5 Degradation of NH4+-N by PCD under different concentrations of SO42- and HCO3- |
| 表 3 不同浓度的SO42-和HCO3-下NH4+-N降解的拟合动力学方程及其分解能效 Table 3 Fitting kinetic equations and energy efficiency for degradation of NH4+-N under different concentrations of SO42- and HCO3- |
由图 5可见, SO42-和HCO3-对NH4+-N的PCD降解存在明显的抑制作用.当模拟废水中SO42-浓度由0 mg·L-1提高到500 mg·L-1, NH4+-N的降解速率迅速降低, 降解速率常数从0.2109 min-1逐渐减少至0.1294 min-1, 分解能效从1.47 g·kW-1·h-1降低到0.87 g·kW-1·h-1, 相应的NH4+-N去除率下降到61.0%.同样地, 随反应体系中HCO3-浓度由0 mg·L-1提高到300 mg·L-1, NH4+-N的降解速率常数和去除率分别下降到0.116 min-1和55.2%, 分解能效降低至0.78 g·kW-1·h-1.产生此现象的原因是, SO42- (姜成春等, 2008)和HCO3-会俘获由O3分解转化的·OH, 转化为对NH4+-N氧化能力较弱的·SO4-和·CO32-, 反应如下:
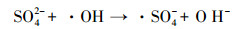
|
(17) |

|
(18) |

|
(19) |
有趣的是, HCO3-对反应的抑制作用要高于SO42-, 原因认为有两方面:其一, HCO3-作为一种典型的·OH的猝灭剂, 对·OH的捕获能力更强(Ma et al., 2000; Busset et al., 2007);其二, HCO3-在碱性环境中会转化为CO2, 对NH4+-N起到一定的吹脱作用.然而, 当废水中SO42-和HCO3-浓度分别提高到600 mg·L-1和360 mg·L-1时, NH4+-N的降解速率常数及其分解能效却没有明显的变化, 原因是在反应周期内由O3转化形成的·OH量是一定的.
4 结论(Conclusions)1) PCD技术因原位产生多种强活性氧化物种, 能实现SCN-的快速氨解, 在不同的pH条件下, SCN-都能在30 min内完全分解, 且碱性条件更有利于反应过程的进行, 当反应体系的pH为10.0时, SCN--N分解的反应速率常数达1.226 min-1.通过对反应体系pH和反应时间的调控, 可控制出水中的NH4+-N和NO3--N的比例, 能为厌氧氨氧化及其配套组合工艺的构建提供条件, 为后续的TN去除提供了水质条件.
2) PCD技术能够氧化不被重铬酸钾(COD分析方法)所氧化的NH4+-N, 氧化产物基本为NO3--N, 此过程几乎检测不出中间产物NO2--N, 随着废水pH的降低以及SO42-和HCO3-浓度的增高均会显著降低PCD对NH4+-N的氧化速率.
Ajo P, Kornev I, Preis S. 2015. Pulsed corona discharge in water treatment:the effect of hydrodynamic conditions on oxidation energy efficiency[J]. Industrial & Engineering Chemistry Research, 54(30): 7452–7458.
|
Bejan D, Graham T, Bunce N J. 2013. Chemical methods for the remediation of ammonia in poultry rearing facilities:A review[J]. Biosystems Engineering, 3: 230–243.
|
Boucabeille C, Bories A, Ollivier P. 1994a. Degradation of thiocyanate by a bacterial coculture[J]. Biotechnology Letters, 16: 425–430.
|
Boucabeille C, Bories A, Ollivier P, et al. 1994b. Microbial degradation of metal complexed cyanides and thiocyanate from mining wastewaters[J]. Environmental Pollution, 84: 59–67.
DOI:10.1016/0269-7491(94)90071-X
|
Busset C, Mazellier P, Sarakha M, et al. 2007. Photochemical generation of carbonate radicals and their reactivity with phenol[J]. Journal of Photochemical Photobiology A:Chemistry, 185: 127–132.
DOI:10.1016/j.jphotochem.2006.04.045
|
Bunce N J, Bejan D. 2011. Mechanism of electrochemical oxidation of ammonia[J]. Electrochemical Acta, 56(24): 8085–8093.
DOI:10.1016/j.electacta.2011.07.078
|
Chen J, Shi H, Lu J. 2007. Electrochemical treatment of ammonia in wastewater by RuO2-IrO2-TiO2/Ti electrodes[J]. Journal of Applied Electrochemistry, 37(10): 1137–1144.
DOI:10.1007/s10800-007-9373-6
|
Dobrin D, Bradu C, Magureanu M, et al. 2013. Degradation of diclofenac in water using a pulsed corona discharge[J]. Chemical Engineering Journal, 234: 389–396.
DOI:10.1016/j.cej.2013.08.114
|
Gerrity D, Stanford B D, Trenholm R A, et al. 2010. An evaluation of a pilot-scale nonthermal plasma advanced oxidation process for trace organic compound degradation[J]. Water Research, 44: 493–504.
DOI:10.1016/j.watres.2009.09.029
|
Gould W D, King M, Mohapatra B R, et al. 2012. A critical review on destruction of thiocyanate in mining effluents[J]. Minerals Engineering, 34: 38–47.
DOI:10.1016/j.mineng.2012.04.009
|
Gupta N, Balomajumder C, Agarwal V K. 2010. Enzymatic mechanism and biochemistry for cyanide degradation[J]. Journal of Hazardous Materials, 176: 1–13.
DOI:10.1016/j.jhazmat.2009.11.038
|
Hoigné J, Bader H. 1983. Rate constants of reactions of ozone with organic and inorganic compounds in water-Ⅱ:Dissociating organic compounds[J]. Water Research, 17: 185–194.
DOI:10.1016/0043-1354(83)90099-4
|
Hoigné J, Bader H, Haag W R, et al. 1985. Rate constants of reactions of ozone with organic and inorganic compounds in water-Ⅲ:Inorganic compounds and radicals[J]. Water Research, 19: 993–1004.
DOI:10.1016/0043-1354(85)90368-9
|
姜成春, 庞素艳, 江进, 等. 2008. 无机阴离子对类-Fenton反应的影响[J]. 环境科学学报, 2008, 28(5): 982–987.
DOI:10.3321/j.issn:0253-2468.2008.05.028 |
Khuntia S, Majumder S K, Ghosh P. 2012. Removal of ammonia from water by ozone microbubbles[J]. Industrial and Engineering Chemistry Research, 52: 318–326.
|
Kim K W, Kim Y J, Kim I T, et al. 2006. Electrochemical conversion characteristics of ammonia to nitrogen[J]. Water Research, 40(7): 1431–1441.
DOI:10.1016/j.watres.2006.01.042
|
Ko C H, Hsieh P H, Chang M W, et al. 2009. Kinetics of pulp mill effluent treatment by ozone-based processes[J]. Journal of Hazardous Materials, 168(2/3): 875–881.
|
Kornev I, Saprykin F, Preis S. 2017. Stability and energy efficiency of pulsed corona discharge in treatment of dispersed high-conductivity aqueous solutions[J]. Journal of Electrostatics, 89: 42–50.
DOI:10.1016/j.elstat.2017.07.001
|
Liu M, Preis S, Kornev I, et al. 2018. Pulsed corona discharge for improving treatability of coking wastewater[J]. Journal of Environmental Science, 64: 306–316.
DOI:10.1016/j.jes.2017.07.003
|
Luo X P, Yan Q, Wang C Y, et al. 2015. Treatment of Ammonia Nitrogen Wastewater in Low Concentration by Two-Stage Ozonization[J]. International Journal of Environmental Research and Public Health, 12(9): 11975–11987.
DOI:10.3390/ijerph120911975
|
Ma J, Graham N. 2000. Degradation of atrazine by manganese-catalysed ozonation-Influence of radical scavengers[J]. Water Research, 34: 3822–3828.
DOI:10.1016/S0043-1354(00)00130-5
|
Malik M A, Ghaffar A, Malik S A. 2001. Water purification by electrical discharges[C]. Plasma Sources Science & Technology, 10: 82-91
|
McKay G, Dong M M, Kleinman J L, et al. 2011. Temperature dependence of the reaction between the hydroxyl radical and organic matter[J]. Environmental Science Technology, 16: 6932–6937.
|
Meunier L, Canonica S, Gunten U. 2006. Implications of sequential use of UV and ozone for drinking water quality[J]. Water Research, 40(9): 1864–1876.
DOI:10.1016/j.watres.2006.02.030
|
Mirat D, Gurol, William M, et al. 1985. Kinetics and mechanism of ozonation of free cyanide species in water[J]. Environmental Science Technology, 19(9): 804–809.
DOI:10.1021/es00139a006
|
Pan J, Wei C, Fu B B, et al. 2018. Simultaneous nitrite and ammonium production in an autotrophic partial denitrification and ammonification of wastewaters containing thiocyanate[J]. Bioresource Technology, 252: 20–27.
DOI:10.1016/j.biortech.2017.12.059
|
Pan J X, Ma J D, Wu H Z, et al. 2018. Simultaneous removal of thiocyanate and nitrogen from wastewater by autotrophic denitritation process[J]. Bioresource Technology, 267: 30–37.
DOI:10.1016/j.biortech.2018.07.014
|
Pan J X, Ma J D, Wu H Z, et al. 2019. Application of metabolic division of labor in simultaneous removal of nitrogen and thiocyanate from wastewater[J]. Water Research, 150: 216–224.
DOI:10.1016/j.watres.2018.11.070
|
Pokryvailo A, Wolf M, Yankelevich Y, et al. 2006. High-power pulsed corona for treatment of pollutants in heterogeneous media[C]. IEEE Transactions on Plasma Science, 34: 1731-1743
https://www.researchgate.net/publication/3166544_High-Power_Pulsed_Corona_for_Treatment_of_Pollutants_in_Heterogeneous_Media |
Ramirez P, Toledo H, Guiliani N, et al. 2002. An exported rhodanese-like protein is induced during growth of acidithiobacillus ferrooxidans in metal sulfides and different sulfur compounds[J]. Applied and Environmental Microbiology, 68: 1837–1845.
DOI:10.1128/AEM.68.4.1837-1845.2002
|
Sharma V K, Yngard R A, Cabelli D E, et al. 2008. Ferrate (Ⅵ) and ferrate (Ⅴ) oxidation of cyanide, thiocyanate, and copper (Ⅰ) cyanide[J]. Radiation Physics and Chemistry, 77(6): 761–767.
DOI:10.1016/j.radphyschem.2007.11.004
|
Sun Y, Guan Y, Wang H, et al. 2019. Autotrophic nitrogen removal in combined nitritation and Anammox systems through intermittent aeration and possible microbial interactions by quorum sensing analysis[J]. Bioresource Technology, 272: 146–155.
DOI:10.1016/j.biortech.2018.10.017
|
Van Leeuwen J, Badriyha B, Vaczi S. 2003. Investigation into ozonation of coal coking processing wastewater for cyanide, thiocyanate and organic removal[J]. Ozone:Science & Engineering, 25: 273–283.
|
韦聪, 何美玲, 韦景悦, 等. 2018. 无汞测试高氯低有机物浓度模拟工业废水COD的原理[J]. 中国测试, 2018, 44(11): 1–11.
DOI:10.11857/j.issn.1674-5124.2018.11.001 |
韦朝海. 2012. 煤化工中焦化废水的污染、控制原理与技术应用[J]. 环境化学, 2012, 31(10): 1465–1472.
|
Young C. 2001. Remediation technologies for the management of aqueous cyanide species[C]. Cyanide: Social, Industrial and Economic Aspects. 175-194
|