 2020, Vol. 40
2020, Vol. 40



2. 中国船舶重工集团公司第七一一研究所, 上海 201108
2. Shanghai Marine Diesel Engine Research Institute, Shanghai 201108
化工生产、汽车制造、交通运输等过程中产生的挥发性有机化合物(VOCs)是造成大气污染的重要原因之一.VOCs主要包括苯系物芳香烃、脂肪烃和氯代烃等, 在空气中易引发光化学烟雾及温室效应, 对自然环境和人体健康有极大危害, 因此VOCs废气的去除方法受到了国内外学者的广泛关注.催化氧化是目前最有效的VOCs废气处理技术, 具有转化率高、无二次污染等优点, 其核心在于高性能催化剂的研发.VOCs催化剂主要包括贵金属催化剂(Pt、Pd等)和过渡金属氧化物催化剂(Cu、Ce、Ni、Mn、Co等).贵金属催化剂有较高的催化活性, 但存在成本高, 易失活, 抗中毒能力差等问题(郑宽等, 2014); 过渡金属氧化物催化剂由于其较低的成本和较强的稳定性而日益受到关注, 其中锰氧化物及铈氧化物获得了广泛的研究.锰氧化物容易形成混合氧化态(Zhang et al., 2013), 具有较高的储氧能力(Li et al., 2014), 常用于甲苯、乙酸乙酯和正己烷的氧化(Kamal et al., 2016; Du et al., 2018).铈氧化物具有丰富的氧空位和较强的Ce3+↔ Ce4+循环转化能力, 同时能大幅提高金属分散性, 对苯、丙酮、氯苯等VOCs气体的氧化起到良好的效果(He et al., 2015; Kamal et al., 2016).但非贵金属氧化物催化剂的低温活性较差, 通常需要达到近300 ℃才能将苯等VOCs气体完全转化(Wang et al., 2013; Deng et al., 2018).
为了进一步提高催化反应去除VOCs废气的效率, 研究者们尝试了许多非常规催化方法.等离子体辅助技术在低温下对VOCs有较高的转化效果(王雪青等, 2019), 但在实际应用中存在能耗高、选择性差、易生成副产物等问题(Vandenbroucke et al., 2011; 刘安琪等, 2019).相关文献研究(Sekine et al., 2011; Oshima et al., 2013; Oshima et al., 2014)提出了一种稳恒直流电场辅助催化反应体系, 并将其应用于乙醇蒸汽重整、甲烷重整和反相水煤气转移反应中, 发现电场可以显著提升催化剂的低温活性, 但对其促进催化活性的原因尚未深入探究, 且目前未有关于电场促进VOCs气体催化氧化过程的研究报道.
本文以VOCs中的典型气体苯为研究对象, 采用自蔓延燃烧法制备了一系列具有导电性的锰铈复合氧化物催化剂, 对其在苯氧化反应中的催化性能进行了评价, 并引入稳恒直流电场辅助技术, 探究电场协同效应对苯催化氧化活性的影响.研究了电场引入前后催化剂性能与理化性质之间的内在关系, 提出电场促进苯催化氧化的反应机理.
2 实验(Experiments) 2.1 催化剂样品制备采用自蔓延燃烧合成法(朱霖等, 2012; Li et al., 2018)制备了锰铈复合氧化物催化剂.首先按照不同的Mn/Ce配比将Ce(NO3)3 · 6H2O及Mn(NO3)2(50%水溶液)溶于去离子水中作为前驱液, 并依据化学计量比加入甘氨酸(C2H5NO2)作为燃料.混合溶液在50 ℃下充分搅拌1 h后转移至坩埚内, 在马弗炉中迅速加热至400 ℃并持续焙烧4 h, 最终将燃烧形成的粉末经压实、研磨后筛分至40~80目颗粒备用.按照不同的Mn、Ce负载量物质的量之比, 将催化剂分别标记为MnxCey.
2.2 催化剂性能评价苯的催化氧化反应在固定床流动反应器内进行, 如图 1所示.将催化剂填充于石英管反应器中, 反应器安装在管式炉内, 炉温由PID控制器调节.采用大连泰斯曼公司的TRC2025P10-1000型可调节电源产生稳恒直流电场, 催化剂两端分别通过不锈钢电极连接导线, 与电源形成闭合回路并获得恒定电流输出.电流及催化剂两端的电压数值可直接由电源发生器中的测量单元读取, 为消除电流热效应产生的温度偏差, 另将一热电偶插入催化剂床层测量反应过程中的实际温度.
 |
| 图 1 稳恒直流电场辅助催化装置示意图 Fig. 1 Schematic description of catalytic experimental equipment combined with steady DC electric field |
在每次测量中使用0.3 mL的催化剂样品, 反应气体由C6H6(0.1%)、O2(5%)组成, 以N2作为载气, 总流量为150 mL · min-1(空速30000 h-1).反应产物成分通过Nicolet 6700型红外光谱仪以1 Hz的采样率实时监测, 待反应器出口成分稳定时记录数据.催化剂活性通过苯的转化率进行衡量, 计算公式如下:
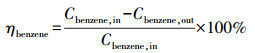
|
(1) |
式中, Cbenzene, in、Cbenzene, out分别为苯在反应器中的进出口浓度.
2.3 催化剂理化性能表征对新鲜催化剂样品和经过电场处理的样品(300 ℃纯N2气氛下经电场处理3 h)的理化性能进行了表征:采用Rigaku D/max-2200/PC型X射线衍射仪进行催化剂物相结构分析(XRD), 扫描角度2θ为20°~90°; 采用JEOL JEM2100透射电子显微镜对催化剂的微观结构进行表征(HRTEM); 采用PHI 5000C ESCA科学电子能谱仪分析催化剂表面元素价态(XPS), 结合能以C1s 284.8 eV为基准, 通过XPS peak 4.1软件进行分峰拟合; 在固定床流动反应器中进行了氢气程序升温还原实验(H2-TPR), 采用GC 2014气相色谱仪和TCD检测器监测氢气含量变化; 采用Nicolet 6700型红外光谱仪, 通过原位漫反射红外傅里叶变换技术(in-situ FTIR)探究苯在催化剂表面的氧化过程(样品先在300 ℃纯N2气氛预处理0.5 h, 实验气氛与性能评价中的设定相同, 测试温度为50~300 ℃).
3 结果与讨论(Results and discussion) 3.1 催化剂性能在电流为0 mA(未施加稳恒直流电场)及5 mA条件下测试了MnxCey催化剂对苯的催化活性, 结果如图 2所示.根据测试结果, 按催化剂中的元素比分别将Ce/Mn比例大于等于1的3种催化剂记为A组, Ce/Mn比例小于1的2种催化剂记为B组, 以Ce为主体的A组催化剂活性明显高于以Mn为主体的B组催化剂.随着Ce/Mn比例从1 : 5上升至3 : 1, 催化效率不断提高, 其中Mn1Ce3表现出最高的活性, 在217.4 ℃可达50%的苯转化效率, 在250.7 ℃可达90%的苯转化效率, Mn1Ce5效率略低于Mn1Ce3.
 |
| 图 2 苯在不同MnxCey催化剂上的转化效率 (注:“EF”表示电场, 实心曲线为无电场条件下的转化效率; 空心曲线为5 mA稳恒直流电场条件下的转化效率.) Fig. 2 Conversion efficiency of benzene over different MnxCey catalysts |
在施加5 mA电流的条件下, B组催化剂的活性提升较小, 而A组催化剂在低温区间内的效率显著提高了30%~60%.随着Ce/Mn比例增大, 电场对催化剂性能促进作用不断增强, 其中Mn1Ce3在通电时仍表现出最高的活性, 其效率达到50%时对应的起燃温度T50低至155.0 ℃, T90低至202.4 ℃, 分别下降了62.4 ℃和48.3 ℃.在所有催化剂测试过程中, 均未检测到CO或O3等副产物, CO2产率与苯转化率呈相同变化趋势.
各组催化剂通电前后的关键转化温度点对比示于表 1, 达到相同转化效率时, A组所需的温度远低于B组, 说明电场的促进作用与催化剂的组分密切相关, Ce的添加可以显著增强电场与催化剂之间的协同效应, Ce含量低时几乎没有协同效应.反应过程中的电压及电场强度与各催化剂的电性能有关, 在不同温度下随着催化剂电阻的改变而变化, 如表 1所示.
| 表 1 苯在不同MnxCey催化剂上氧化的实验结果 Table 1 Test results of benzene oxidation over different MnxCey catalysts |
图 3为MnxCey催化剂在未通电和电场作用下反应的阿伦尼乌斯分布图, 对应的表观活化能Ea示于表 1中.A、B两组催化剂的活化能有明显差异, A组催化剂引入电场后反应活化能明显降低, 其中Mn1Ce3催化剂在电场中的反应活化能比无电场时降低了20.01 kJ · mol-1.
 |
| 图 3 苯在不同MnxCey催化剂上反应的阿伦尼乌斯图 (a.无电场反应; b.稳恒直流电场协同反应) Fig. 3 Arrhenius plots of benzene oxidation over different MnxCey catalysts (a. reaction without electric field; b. reaction cooperate with electric field) |
MnxCey催化剂的XRD谱图如图 4a所示.2θ为28.55°、33.08°、47.49°、56.34°、59.09°、69.42°、79.70°、79.08°、88.43°的衍射峰分别对应于CeO2晶相(JCPDS 89-8436), 2θ为36.09°、44.42°、50.77°、53.87°、64.63°的衍射峰分别对应于Mn3O4晶相(JCPDS 89-4837).在锰含量较低的A组催化剂中, 所有衍射峰都对应于立方尖晶石结构的CeO2物相, 未发现锰氧化物衍射峰, 说明Mn在CeO2中分散良好, 未发生MnOx的团聚现象.随着锰含量的增加, 衍射峰变宽, 强度减弱, 同时CeO2在(111)晶面衍射峰对应的衍射角2θ=28.55°处发生偏移, 说明一定数量的锰离子进入CeO2晶格中, 形成Mn-Ce-O固溶体, 抑制CeO2的结晶形成, 形成晶体缺陷, 有利于提高催化活性(Deng et al., 2018).
 |
| 图 4 不同MnxCey催化剂的XRD谱图 (a.未经电场处理的样品, b.电场处理后的样品) Fig. 4 XRD patterns of different MnxCey catalysts (a. fresh samples; b. samples treated by electric filed) |
在B组催化剂的衍射图样中同时观测到了Mn3O4及CeO2两种物相, 由于Ce3+(0.134 nm)及Ce4+(0.114 nm)的离子半径远大于Mn3+(0.064 nm)及Mn4+(0.060 nm)(Zhao et al., 2019), 铈离子无法进入Mn3O4晶格中, 部分锰离子进入了CeO2晶格.当锰离子含量超过了其在CeO2中的溶解极限时(Kang et al., 2006), 过量的锰在CeO2表面以Mn3O4相聚集, 因此, Mn1Ce3催化剂拥有最高的催化活性, 可能与其较高的锰离子分散度有关.
如图 4b所示, 电场处理后催化剂XRD谱图中衍射峰的强度和位置无明显变化, 说明电场不会改变催化剂的晶体结构, 电场对催化活性的提升作用可能是诱导锰、铈离子发生电子迁移等因素导致, 但无法由XRD方法检测到, 需通过下文中的X射线能谱等方法进行检测.
3.2.2 HRTEM表征结果取Mn5Ce1、Mn1Ce1、Mn1Ce5新鲜样品及通电处理后的样品进行HRTEM测试.如图 5所示, 新鲜Mn1Ce5和Mn1Ce1图像中测得间距为0.312 nm、0.189 nm的晶格条纹, 分别对应CeO2的(111)和(220)晶面, 未发现锰氧化物的晶格条纹, 说明锰在CeO2中分散良好.而在Mn5Ce1图像中除了存在CeO2条纹, 还有大量间距为0.249 nm和少量间距为0.471 nm的晶格条纹, 分别对应于Mn3O4的(211)晶面和Mn2O3的(200)晶面.
 |
| 图 5 不同MnxCey催化剂的HRTEM图像 (a.未经电场处理的Mn5Ce1; b.未经电场处理的Mn1Ce1; c.未经电场处理的Mn1Ce5; d.电场处理后的Mn5Ce1; e.电场处理后的Mn1Ce1; f.电场处理后的Mn1Ce5) Fig. 5 HRTEM images of different MnxCey catalysts (a. fresh Mn5Ce1; b, fresh Mn1Ce1; c. fresh Mn1Ce5; d. Mn5Ce1 treated by electric filed; e. Mn1Ce1 treated by electric filed; f. Mn1Ce5 treated by electric filed) |
通电处理后, Mn1Ce5和Mn1Ce1图像中出现了晶格间距为0.293 nm和0.162 nm的新条纹, 分别归属于Ce2O3的(011)晶面和MnO2的(102)晶面.说明在电场中可能发生了CeO2向Ce2O3的还原以及锰离子价态的变化, 而在通电处理后的Mn5Ce1中未检测到相应条纹, 其晶格条纹形貌与原样品基本一致, 根据B组催化剂在电场中活性提升较小的实验结果, 说明电场协同效应可能与锰铈离子价态的变化有关.
3.2.3 XPS表征结果对新鲜MnxCey催化剂样品及通电处理后的样品进行XPS表征分析, 获得的表面原子组成及不同价态元素相对含量示于表 2中.由Mn2p和Ce3d谱峰强度分别测得催化剂表面锰与铈元素的总量, 以Mn/(Mn+Ce)衡量锰元素含量, 并与体相计量值进行比较, 如图 6所示.当锰含量为25%时(对应Mn1Ce3), 表面Mn/(Mn+Ce)与体相计量值相等, 说明锰离子在基体和表面呈均匀分布.随着锰含量的增加, 表面Mn/(Mn+Ce)比例明显大于体相中的比例, 此时大量锰离子集中堆积分布于表面, 与XRD中的结果相符.
| 表 2 电场处理前后不同MnxCey催化剂表面的原子组成 Table 2 Surface atomic composition of fresh and treated MnxCey |
 |
| 图 6 不同MnxCey催化剂表面Mn元素含量与体相Mn元素含量对比 Fig. 6 Mn content distribution in different MnxCey catalysts |
MnxCey催化剂通电处理前后的Mn2p谱图如图 7a~7b所示.结合能位于642.1 eV和653.8 eV的两个特征峰分别对应于Mn2p 3/2和Mn2p 1/2分裂自旋轨道, 其中Mn2p 3/2宽峰可进一步分解为两个峰, 结合能分别为642.7~643.8 eV和641.2~641.5 eV(Zeng et al., 2015), 分别对应于Mn4+和Mn3+.在MnxCey催化剂XRD谱图中未发现MnO2微晶相, 但在XPS中检测到了不同含量的Mn4+, 说明催化剂中存在非晶相的不定型MnO2.
 |
| 图 7 不同MnxCey催化剂的XPS谱图 (a.新鲜样品的Mn2p谱图; b.电场处理后样品的Mn2p谱图; c.新鲜样品的Ce3d谱图; d.电场处理后样品的Ce3d谱图; e.新鲜样品的O1s谱图; f.电场处理后样品的O1s谱图) Fig. 7 XPS spectra of different MnxCey catalysts (a. Mn2p spectra of fresh MnxCey; b. Mn2p spectra of treated MnxCey; c. Ce3d spectra of fresh MnxCey; d. Ce3d spectra of treated MnxCey; e. O1s spectra of fresh MnxCey; f. O1s spectra of treated MnxCey |
Mn4+浓度随Ce/Mn比的升高逐渐增大, 且其浓度变化趋势与催化效率的变化趋势一致, A组催化剂中的Mn4+浓度明显高于B组, 较高的Mn4+浓度可能是提高催化剂性能的主要原因之一.
许多研究表明(Craciun et al., 1998; Machocki et al., 2004; Zhang et al., 2013), Mn4+在MnOx催化氧化反应中作为活性位点, 较高的Mn4+浓度能带来更好的催化效果.A组催化剂经电场处理后, Mn4+浓度明显提升, 进一步说明Mn4+浓度对催化性能的影响.Mn4+浓度提升的同时Mn3+浓度相应下降, 说明电场中Mn4+的增加源于Mn3+的氧化.
如图 7c~7d所示, Ce3d谱图主要由Ce3d 3/2和Ce3d 5/2电子态对应的两组峰构成, 其中结合能位于882.0、888.85、898.4、901.05、907.45和916.7 eV的特征峰归属于Ce4+, 结合能位于883.5、898.9、904.05 eV的特征峰归属于Ce3+ (Paparazzo, 2010).随着Ce/Mn比的增大, Ce3+浓度升高, Ce3+浓度的变化趋势与催化效率的变化趋势一致, 其中活性最佳的Mn1Ce3催化剂样品中Ce3+浓度最高.研究表明(Feng et al., 2019; Guo et al., 2019), Ce3+的存在易使催化剂表面形成电荷不平衡和氧空位缺陷, 促进晶格氧的转移和释放, 有利于提高催化性能.
结合锰和铈的XPS谱图发现表面Mn4+和Ce3+浓度在各催化剂中始终呈同步变化趋势, 说明在催化剂制备过程中可能存在部分Ce4+、Mn3+与Ce3+、Mn4+之间的相互转化, 即Ce4++ Mn3+↔ Ce3++ Mn4+氧化还原过程, 体现了两种元素较强的协同作用.
电场处理后A组催化剂中的Ce4+浓度明显降低, B组催化剂中Ce4+浓度变化不大, 说明电场可能诱导以CeOx结构为主的A组催化剂发生Ce4+的还原, 而以MnOx结构为主的B组催化剂在电场下发生Ce4+还原反应的可能性较低.这是由于在氧化物半导体中, 尖晶石结构的导电是通过氧空位的生成和氧离子对氧空位的占据来实现的, 所以CeO2在电场中容易失去氧离子, 并发生价态下降.
O1s XPS谱线如图 7e~7f所示, 根据表面氧来源种类的不同, 可将其分解为表面晶格氧、表面吸附氧(O22-, O-, OH-等含氧基团)及表面吸附水分子中的氧, 分别记为OL、OA、OW, 结合能分别对应于529.4、531.0、532.3 eV(Li et al., 2016).从氧的XPS谱线可以观察到催化剂表面水分子中的氧含量几乎可以忽略, 氧物种主要为晶格氧和吸附氧, 其中, 表面吸附氧具有比晶格氧更高的转移能力和反应活性, 对催化氧化过程起着至关重要的作用(Santos et al., 2010; Zhao et al., 2019).
电场处理后, 催化剂中表面吸附氧OA所占的比例均有不同程度的上升, 由于通电处理时催化剂处于纯N2气氛中, 没有气相氧的补充, 因此电场下表面吸附氧的增加仅可能由金属氧化物载体中的晶格氧OL转变而来.结合Mn、Ce价态变化分析, 电场可以促进Ce4++ Mn3+↔ Ce3++ Mn4+正向反应, 导致生成更多的活性位点Mn4+, 且在Ce4+向Ce3+的还原的同时发生了晶格氧向表面吸附氧的转化, 提高了催化氧化活性.
3.2.4 H2-TPR表征结果为了探究MnxCey催化剂的还原性, 以CeOx、MnOx作为参考样本, 对未通电及电场作用下的样品分别进行了H2-TPR测试, 归一化结果如图 8所示.纯锰氧化物的谱图由峰值位于215 ℃(α峰)及423 ℃(β峰)的两个还原峰组成, 分别为Mn4+及Mn3+对应氧化物的还原; 纯铈氧化物的谱图由峰值位于252 ℃(λ峰)、477 ℃(μ峰)及765 ℃(γ峰)的3个还原峰组成, 分别对应于CeO2表面、下表面及体相中氧物种的还原(Feng et al., 2019).与纯氧化物相比, 锰铈催化剂的还原性明显提高.B组催化剂中锰氧化物的两个还原峰起始于较高温度, 未观测到关于Ce的还原, 可能由于CeO2含量较少且表面有大量的锰氧化物团聚, 导致还原性的降低.在A组催化剂中则出现了关于Mn和Ce的还原峰, 由于锰铈氧化物在低温段的还原温度接近, 还原峰在部分区间重叠, 难以完全分离.A组催化剂的还原谱图主要包括4个峰, 从低温起第1个峰为CeO2表面氧物种的还原峰与Mn4+还原峰的叠加, 第2个峰为Mn3+的还原, 第3个峰为CeO2下表面氧物种的还原, 最后一个谱峰对应体相中CeO2氧物种的还原, 其中高温段的两个CeO2还原峰(μ、γ峰)在A组催化剂中的位置基本相同, 但Mn1Ce3在低温段对应于表面CeO2(λ峰)的还原温度明显低于Mn1Ce5及Mn1Ce1.随着Ce/Mn比例从1 : 5上升至3 : 1, 可观察到Mn4+(α峰)及Mn3+(β峰)的还原温度逐渐降低, 催化剂对应的起始还原温度也不断降低, Mn1Ce5相对于Mn1Ce3的各个还原峰温度略有提高.Mn1Ce3的还原温度最低, 说明其所含氧物种的还原能力最强, 对应最高的催化活性.
 |
| 图 8 不同MnxCey催化剂的H2-TPR谱图 (a.无电场反应; b.稳恒直流电场协同反应)(注:图 8a中的曲线以虚线表示于图 8b中作为对比参考) Fig. 8 H2-TPR patterns of different MnxCey catalysts (a. reaction without electric field; b. reaction cooperate with electric field) |
电场中, B组催化剂的两个还原峰温度都略有降低, 峰型基本不变, 说明电场对B组催化剂中Mn4+及Mn3+的含量和还原能力未产生影响.而电场中A组催化剂对应CeO2、Mn4+、Mn3+氧化物的还原温度均有明显降低, 且可观察到电场中Mn4+氧化物还原峰对应的H2消耗量显著增加, Mn3+氧化物还原峰消耗的H2量相应减少, 这与XPS结果吻合, 表明Ce4++ Mn3+↔ Ce3++ Mn4+反应会在电场中发生, 导致A组催化剂中形成更多的Mn4+、Ce3+及活性氧物种, 催化剂还原性增强, 有助于提高在电场中对苯的催化氧化活性.
3.2.5 in-situ FTIR表征结果取催化性能最佳的Mn1Ce3催化剂进行原位红外表征, 探究苯在催化剂表面的氧化反应过程.无电场氧化反应的探测结果如图 9a所示, 位于1040、1480、1820、1968及3050 cm-1的特征峰来自于苯环中C—H键及碳碳键的变形振动(Shen et al., 2015).在50 ℃及100 ℃仅观测到苯的特征峰, 而在150 ℃出现了位于1568 cm-1的酚类特征峰(Yang et al., 2019), 说明苯可能首先被氧化为酚类物质.随着温度升高, 苯特征峰的强度减弱, 在200 ℃观测到邻苯醌(1403 cm-1)的存在, 位于1313、1418 cm-1的特征峰归因于马来酸物种的烯烃振动(Zeng et al., 2015), 同时存在乙酸物种对应于1366 cm-1的特征峰及少量位于2340 cm-1的CO2特征峰.温度高于250 ℃时, 苯及其中间产物的特征峰大幅减弱, 伴随大量CO2生成.位于1130 cm-1的特征峰归因于气态氧的吸附(O2-)(Li et al., 2019), 在150 ℃以上才被检测到, 说明气态氧在150 ℃以下的吸附速率缓慢, 然而, 在100 ℃时就开始有部分苯被氧化为中间产物, 此时活性氧来源于催化剂中的晶格氧.
 |
| 图 9 苯在不同MnxCey催化剂上氧化的in-situ FTIR谱图 (a.无电场反应; b.稳恒直流电场协同反应) Fig. 9 in-situ FTIR spectra of benzene oxidation over different MnxCey catalysts (a. reaction without electric field; b. reaction cooperate with electric field) |
电场中苯在Mn1Ce3表面氧化反应的in-situ FTIR谱图如图 9b所示.在电场中, 苯的催化氧化过程加快, 中间产物的特征峰在更低的温度下出现, 同时也以更快的速度分解, 在较低的温度下就产生了CO2.由于电场中晶格氧向表面活性氧的转化速度加快, 需要消耗更多的气相氧填充晶格中氧空位, 电场反应中的气态氧吸附峰强度低于无电场反应中的谱峰.值得注意的是, 无电场氧化反应的谱图中, 在800 ~1000 cm-1及1645 cm-1处有明显的特征峰, 其归属于MnO(OH)中的羟基(Aitchison et al., 2005; Li et al., 2009), 表明在苯催化氧化的过程中有部分OH基团会与高活性的MnOx反应生成MnO(OH), 抑制活性位点再生, 降低催化剂的反应活性.而在电场条件下没有观测到羟基的存在, 说明电场可能促进Ce4+与MnO(OH)反应, 加速了催化剂表面的去羟基过程, 使Mn4+活性位点再生的同时释放晶体中的晶格氧.
3.3 电场协同反应机理根据以上实验结果, 可推测苯在MnxCey催化剂上的氧化反应机理如图 10所示, 苯在活性位点上首先转化为酚类物质, 酚类物质进一步被氧化为邻苯醌.活性氧及催化剂的作用使邻苯醌中的苯环形成开口, 产生马来酸及乙酸类中间产物, 并迅速被氧化为水和二氧化碳.
 |
| 图 10 电场协同MnxCey催化氧化苯的反应机理 (a.苯的氧化路径; b.无电场条件下的反应模型; c.稳恒直流电场协同反应模型) Fig. 10 Reaction mechanism of benzene oxidation over MnxCey assisted by electric field (a. oxidation path of benzene; b. reaction model without electric field; c. reaction model cooperate with electric field) |
一般认为苯在催化剂上的氧化过程遵循Mars-Van Krevelen模型(Santos et al., 2010; Li et al., 2014; Zhang et al., 2018), 即苯吸附在催化剂表面活性位点上, 与催化剂表面的活性氧发生反应, 催化剂被还原, 产生的氧空位再由气相氧补充.根据XPS表征中的分析, Mn4+是MnxCey催化剂表面的主要活性位点, 在传统催化方式中, 苯在Mn4+活性位点上被表面活性氧氧化, 同时Mn4+被还原为Mn3+, 再由气相氧将Mn3+重新氧化再生.CeO2作为锰铈固溶体中的储氧材料, 在传统催化方式中主要对氧的吸附转移起促进作用.Mn4+活性位点的高分散性以及催化剂较强的还原性和氧迁移能力是MnxCey催化剂具有较高活性的原因.
电场协同效应下苯的氧化路径与传统催化方式中的路径相同, 但电场显著加快了中间产物的生成转化速度, 并促进了催化剂表面的去羟基过程.此外, 施加电场后催化剂中Mn4+、Ce3+及表面活性氧OA的含量都有一定增加, 说明电场通过提供自由电子促进Ce4+的还原促进了锰铈之间Ce4++ Mn3+↔ Ce3++ Mn4+正向反应, 将Mn3+重新氧化为Mn4+, 不断产生新的活性位点; Ce3+带来的氧空位由氧气补充, 形成锰铈之间的快速氧化还原循环; 同时, 在Ce4+向Ce3+转化过程中促进了晶格氧向表面活性氧的转化, 产生更多表面活性氧, 显著提升MnxCey对苯的催化氧化性能.
4 结论(Conclusions)稳恒直流电场与MnxCey催化剂间的协同效应能显著提高催化剂活性, 降低苯催化氧化反应温度和反应活化能, 其中Mn1Ce3催化剂在电场中催化反应的T50和T90分别降低了62.4 ℃和48.3 ℃, 活化能降低了20.01 kJ · mol-1; 电场协同催化方法能耗低, 去除效率高, 且无副产物形成, 是一种极具发展前景的VOCs废气处理技术.电场可促进MnxCey催化剂表面的去羟基过程及Ce4++ Mn3+↔ Ce3++ Mn4+氧化还原循环, 两者之间的协同效应与活性位点的快速持续再生及晶格氧向表面活性氧的快速转化有关.下一步工作将对电场协同催化在混合VOCs气体中的应用及催化剂稳定性展开研究; 此外, 将基于本文提出的协同催化机理, 开发协同效应更强的催化剂体系, 并尝试脉冲电流等方法进一步降低能耗, 提高VOCs废气去除效率.
Aitchison P, Ammundsen B, Rozière J, et al. 2005. Local structure and lithium-proton ion exchange in Li1.33-x/3CoxMn1.67-2x/3O4 spinels[J]. Solid State Ionics, 176: 813-821. |
Craciun R. 1998. Structure/activity correlation for unpromoted and CeO2-promoted MnO2/SiO2 catalysts[J]. Catalysis Letters, 55(1): 25-31. |
Deng L, Ding Y P, Duan B Q, et al. 2018. Catalytic deep combustion characteristics of benzene over cobalt doped Mn-Ce solid solution catalysts at lower temperatures[J]. Molecular Catalysis, 446: 72-80. |
Du J P, Qu Z P, Dong C, et al. 2018. Low-temperature abatement of toluene over Mn-Ce oxides catalysts synthesized by a modified hydrothermal approach[J]. Applied Surface Science, 433: 1025-1035. |
Feng Z T, Ren Q M, Peng R S, et al. 2019. Effect of CeO2 morphologies on toluene catalytic combustion[J]. Catalysis Today, 332: 177-182. |
Guo Y L, Gao Y J, Li X, et al. 2019. Catalytic benzene oxidation by biogenic Pd nanoparticles over 3D-ordered mesoporous CeO2[J]. Chemical Engineering Journal, 362: 41-52. |
He C, Xu B T, Shi J W, et al. 2015. Catalytic destruction of chlorobenzene over mesoporous ACeOx (A=Co, Cu, Fe, Mn, or Zr) composites prepared by inorganic metal precursor spontaneous precipitation[J]. Fuel Processing Technology, 130: 179-187. |
Kamal M S, Razzak S A, Hossain M M. 2016. Catalytic oxidation of volatile organic compounds (VOCs)-A review[J]. Atmospheric Environment, 140: 117-134. |
Kang C, Kusaba H, Yahiro H, et al. 2006. Preparation, characterization and electrical property of Mn-doped ceria-based oxides[J]. Solid State Ionics, 177: 1799-1802. |
Li B, Chen Y W, Li L, et al. 2016. Reaction kinetics and mechanism of benzene combustion over the NiMnO3/CeO2/Cordierite catalyst[J]. Journal of Molecular Catalysis A:Chemical, 415: 160-167. |
Li B, Huang Q, Yan X K, et al. 2014. Low-temperature catalytic combustion of benzene over Ni-Mn/CeO2/cordierite catalysts[J]. Journal of Industrial and Engineering Chemistry, 20: 2359-2363. |
Li J H, Liang X, Xu S C, et al. 2009. Catalytic performance of manganese cobalt oxides on methane combustion at low temperature[J]. Applied Catalysis B:Environmental, 90: 307-312. |
Li K, Liu K, Ni H, et al. 2018. Electric field promoted ultra-lean methane oxidation over Pd-Ce-Zr catalysts at low temperature[J]. Molecular Catalysis, 459: 78-88. |
Li K, Liu K, Xu D J, et al. 2019. Lean methane oxidation over Co3O4/Ce0.75Zr0.25 catalysts at low-temperature:Synergetic effect of catalysis and electric field[J]. Chemical Engineering Journal, 369: 660-671. |
刘安琪, 李建军. 2019. 低温等离子体协同催化降解VOCs的研究进展[J]. 化工技术与开发, 48(6): 29-32. |
Machocki A, Ioannides T, Stasinska B, et al. 2004. Manganese-lanthanum oxides modified with silver for the catalytic combustion of methane[J]. Journal of Catalysis, 227: 282-296. |
Oshima K, Shinagawa T, Haraguchi M, et al. 2013. Low temperature hydrogen production by catalytic steam reforming of methane in an electric field[J]. International Journal of Hydrogen Energy, 38: 3003-3011. |
Oshima K, Shinagawa T, Nogami Y, et al. 2014. Low temperature catalytic reverse water gas shift reaction assisted by an electric field[J]. Catalysis Today, 232: 27-32. |
Paparazzo E. 2010. Preparation and Characterization of Ce/N-Codoped TiO2 Particles for production of H2 by photocatalytic splitting water under visible light by sun et al. (Catal Lett (2010) 135:219)[J]. Catalysis Letters, 140: 147-150. |
Santos V P, Pereira M F R, Órfão J J M, et al. 2010. The role of lattice oxygen on the activity of manganese oxides towards the oxidation of volatile organic compounds[J]. Applied Catalysis B:Environmental, 99: 353-363. |
Sekine Y, Haraguchi M, Matsukata M, et al. 2011. Low temperature steam reforming of methane over metal catalyst supported on CexZr1-xO2 in an electric field[J]. Catalysis Today, 171: 116-125. |
Shen Y, Wang L F, Wu Y B, et al. 2015. Facile solvothermal synthesis of MnFe2O4 hollow nanospheres and their photocatalytic degradation of benzene investigated by in situ FTIR[J]. Catalysis Communications, 68: 11-14. |
Vandenbroucke A M, Morent R, De Geyter N, et al. 2011. Non-thermal plasmas for non-catalytic and catalytic VOC abatement[J]. Journal of Hazardous materials, 195: 30-54. |
王雪青, 黎欢毅, 王邦芬, 等. 2019. 等离子体场内CeO2催化降解甲醇的表面活性氧物种来源与作用研究[J]. 环境科学学报, 39(8): 2725-2734. |
Wang Z, Shen G L, Li J Q, et al. 2013. Catalytic removal of benzene over CeO2-MnOx composite oxides prepared by hydrothermal method[J]. Applied Catalysis B:Environmental, 138-139: 253-259. |
Yang K, Liu Y X, Deng J G, et al. 2019. Three-dimensionally ordered mesoporous iron oxide-supported single-atom platinum:Highly active catalysts for benzene combustion[J]. Applied Catalysis B:Environmental, 244: 650-659. |
Zeng J L, Liu X L, Wang J, et al. 2015. Catalytic oxidation of benzene over MnOx/TiO2 catalysts and the mechanism study[J]. Journal of Molecular Catalysis A:Chemical, 408: 221-227. |
Zhang X L, Ye J h, Yuan J, et al. 2018. Excellent low-temperature catalytic performance of nanosheet Co-Mn oxides for total benzene oxidation[J]. Applied Catalysis A:General, 566: 104-112. |
Zhang Y G, Qin Z F, Wang G F, et al. 2013. Catalytic performance of MnOx-NiO composite oxide in lean methane combustion at low temperature[J]. Applied Catalysis B:Environmental, 129: 172-181. |
Zhao L L, Zhang Z P, Li Y S, et al. 2019. Synthesis of CeaMnOx hollow microsphere with hierarchical structure and its excellent catalytic performance for toluene combustion[J]. Applied Catalysis B:Environmental, 245: 502-512. |
郑宽, 付名利, 吴军良, 等. 2014. 氧化甲苯的MnOx-CeO2催化剂表面活性物种研究[J]. 环境科学学报, 34(11): 2885-2891. |
朱霖, 林赫, 管斌, 等. 2012. SHS方法制备的锰铁系NH3-SCR催化剂的性能研究和机理分析[J]. 车用发动机, 3: 72-77. |