 2020, Vol. 40
2020, Vol. 40



2. 南京工业大学环境学院, 南京 211816
2. School of Environmental Science and Engineering, Nanjing Tech University, Nanjing 211816
垃圾焚烧技术凭借减量化、资源化和无害化的优势, 得到了丰富的应用.但垃圾焚烧后产生的二次污染仍然不容小觑, 特别是所排烟气中的氮氧化物(NOx)和二英, 成为垃圾焚烧技术发展的一大拦路虎(付建平等, 2017).一方面, 二英作为剧毒污染物, 严重污染环境和危害人类健康, 因此对二英污染采取相关的防治措施已迫在眉睫(李雁等, 2019).另一方面, 高浓度的NOx不仅能与水蒸气结合形成硝酸(HNO3)、酸雨(Xu et al., 2018), 还会与有机挥发性污染物(VOCs)产生光化学反应, 在强光辐射下生成光化学烟雾, 极大危害人类眼睛以及呼吸道, 甚至会导致人类患上癌症(Jin et al., 2019).随着相关法律法规的完善和加强, 常规污染物的排放标准日渐提高, 二英等痕量有机污染物也被归入监测范围.
针对NOx的脱除, 最具代表性的是干法脱硝中的选择性催化还原技术, 即SCR技术(Ning et al., 2015).该技术也是最早在燃料电厂中被使用的, 经过长时间的发展和完善, 现已成为控制NOx排放的主流技术, 常见的脱硝催化剂主要分为金属氧化物、分子筛以及贵金属催化剂(Liu et al., 2019).针对二英的脱除, 常见的有催化氧化法(Parizek et al., 2008; Liu et al., 2015; Gan et al., 2018)、吸附剂吸附法(Wang et al., 2017; Cui et al., 2017)、紫外光催化(Niu et al., 2015)和低温等离子技术(Yoshida et al., 2009).其中最为高效且有望成功商业化的方法便是金属氧化物催化剂催化脱除, 虽然降解机理仍然不能被清晰且深层次地解释, 但大致有两种方式可以从根本上实现降解, 一种是脱氯, 另一种是苯环断裂.与此同时, 氧化钒作为常见脱硝催化剂的活性组分, 不仅具有良好的氧化还原性能, 且钒基脱硝催化剂还具有较强的表面酸性, 能够有效保证催化剂的脱硝活性(Xu et al., 2019).MnOx则因为其多种价态的转换而具有良好的低温脱硝活性(Chen et al., 2019), 因此VOx和MnOx活性组分的复合可以使得催化剂在中低温区间具有优异的脱硝活性和催化分解二英能力.本实验通过浸渍法制备了VOx/TiO2、MnOx/TiO2以及VOx-MnOx/TiO2等多种催化剂, 并对其在200~300 ℃温度区间下脱硝和脱除二英的效果进行了研究, 从而研制出适合处理垃圾焚烧等工业废气的协同脱硝脱二英催化剂.
2 实验部分(Experimental) 2.1 催化剂的制备量取50 mL去离子水, 再按5%的负载量依次加入偏钒酸铵和四水合乙酸锰(MnO2和V2O5的物质的量的比分别为6 : 0、5 : 1、4 : 1、3 : 1、2 : 1以及0 : 6), 利用恒温水浴锅将溶液加热到80 ℃使其成为均一透明溶液后加入1 g TiO2;接着将样品恒温搅拌3 h, 搅拌速度设置为400 r · min-1, 其中25 ℃搅拌2 h, 85 ℃搅拌1 h;然后将样品放入烘箱中120 ℃下干燥10 h, 紧接着放置于一体式马弗炉中在550 ℃焙烧5 h.最后取出焙烧完成的催化剂样品并将其研磨过筛得到40~60目的催化剂颗粒, 其中样品根据MnO2和V2O5的物质的量的比分别缩写为0V6M/T、1V5M/T、1V4M/T、1V3M/T、1V2M/T和6V0M/T.
2.2 催化剂单独活性测试脱硝催化评价装置简图如图 1所示, 取1 mL催化剂颗粒置于内径为1 cm的石英管内, 石英管置于催化剂评价装置右侧的加热炉内, 温控仪控制加热时间与反应温度.模拟配气成分(体积比)为800×10-6 NO、800×10-6 NH3、600×10-6 SO2, 11%O2, 载气为N2.从50 ℃开始测试, 每隔50 ℃采集一次出口气体, 直至温度升到300 ℃为止, 用烟气分析仪(MRU VarioPlus, Germany)对所采集的气体进行分析.NO转化率的计算公式见式(1):
 |
| 图 1 催化评价装置图 Fig. 1 Catalytic evaluation device diagram |
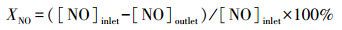
|
(1) |
催化降解二英采用气相色谱仪(GC1024, 岛津)进行分析研究.选用邻二氯苯代替二英来进行测试, 原因是:二英毒性大、结构复杂且价格昂贵不宜研究, 而邻二氯苯具有和二英相似的分子结构, 同时邻二氯苯相较于二英来说毒性较小、挥发性更高且价格便宜.不同于脱硝测试的是, 脱除二英测试需将微量注射泵和恒温油浴锅接入催化评价装置.模拟配气成分(体积比)为11%O2, 300×10-6邻二氯苯, 其余为N2.从200 ℃开始测试, 每隔50 ℃采集一次出口气体, 直至温度升到300 ℃为止.当达到相应的温度后, 用微量注射器抽取集气袋内气体注射入气相色谱仪, 用气相色谱仪对催化分解二英的效果进行分析.在测试之前会分别测试200、250和300 ℃时不加催化剂的邻二氯苯的基准峰面积, 测试结果用公式(2)表示(A0:基准峰面积;A:测试峰面积):
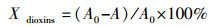
|
(2) |
催化剂的晶型结构分析采用X射线衍射仪(Rigaku D max/RB型, 日本理学公司), 测试条件为5°~90°, 扫描速度为10° · min-1.催化剂的X射线光电子能谱(XPS)测试采用AXIS ULTRA DLD型仪器, 催化剂在测试前需预处理(100 ℃下干燥24 h).催化剂的氧化还原性能采用氢气程序升温还原(H2-TPR)法进行分析表征.测试仪器为:Micromeritics TPD / TPR 2900.测试条件如下:温度范围为50~800 ℃, 升温速率为5 ℃ · min-1, 样品用量为5 mg, 气体组成为5%H2 + 95%Ar, 气体流量为50 mL · min-1.在还原实验前, 将样品在氩气下加热至400 ℃, 然后冷却至100 ℃.采用美国Micromeritics公司的ASAP 2020M V3.00H型比表面积及微孔分析仪对催化剂比表面积、孔容孔径进行测试, 催化剂在测试前需进行预处理(350 ℃真空处理3 h, N2为吸附质).通过Nicolet is50光谱仪收集原位漫反射红外傅里叶变换光谱(原位DRIFTS).将催化剂在300 ℃下在N2气流下预热2 h, 然后冷却至所需温度.对于NH3与预吸附NO + O2之间的反应, 将600×10-6 NO + 5%O2通入系统30 min.在温度升至250 ℃后, 停止通入NO + O2并开始将600×10-6 NH3通入系统.在不同时间收集原位DRIFT光谱.对于NO + O2与预吸附NH3之间的反应, 气体的顺序是反向的, 但步骤与NH3和预吸附的NO + O2之间的反应相似.
3 结果与讨论(Results and discussion) 3.1 催化性能图 2a为不同组分催化剂在不同温度下的脱硝效率.可以看出, 在200~300 ℃这一温度区间里, 当温度从250 ℃升温至300 ℃时, 只有6V0M/T和1V2M/T催化剂脱硝效率在继续上升, 其它4个催化剂脱硝效率都略有下降.1V5M/T催化剂在200 ℃和250 ℃时脱硝效果最好, 6V0M/T催化剂脱硝效率最差.另外, 可以看出, 随着MnOx含量的增加, 催化剂的低温催化效率逐渐增加.但由纯MnOx组成的0V6M/T催化剂其低温脱硝效率不如1V5M/T和1V4M/T.由此可以看出, 钒钛催化剂中掺入适量MnOx可以有效改善其低温脱硝性能, 其脱硝活性温度区间向200~300 ℃偏移.这主要是由于MnOx和VOx都是高效的脱硝活性组分, 而1V5M/T和1V4M/T等催化剂的性能优于纯钒钛催化剂以及纯锰钛催化剂, 说明MnOx和V2O5具有一定的协同催化作用(Zhao et al., 2015).
 |
| 图 2 反应温度对NO(a)和邻二氯苯(b)催化活性的影响 Fig. 2 Effect of reaction temperature on catalytic activity of NO(a) and O-dichlorobenzene(b) |
图 2b为不同组分催化剂在不同温度下的邻二氯苯脱除效率.可以看出, 在200~300 ℃这一区间里, 催化剂的邻二氯苯脱除效率随着反应温度的升高而逐渐增加, 6V0M/T催化剂在300 ℃时脱除邻二氯苯的效果最好, 0V6M/T催化剂效率最差, 即钒基催化剂脱除邻二氯苯的效果优于锰基催化剂的.钒基催化剂中掺入MnO2会轻微降低催化剂的邻二氯苯脱除效率.综合比较6种催化剂的脱硝和脱邻二氯苯效率, 选择1V3M/T催化剂作为最优催化剂, 主要是因为其在200~300 ℃温度区间内邻二氯苯效率只是略微低于0V6M/T催化剂, 而脱硝效率明显提高.另外, 在实际生产中, V2O5含量的减少也意味着催化剂生产成本的降低.
3.2 物理性质分析分析了不同催化剂的X射线衍射图谱, 结果如图 3所示.所有催化剂的衍射图谱都对应于锐钛矿TiO2(2θ= 25.3°、37.8°、48.0°、53.9°、55.1°)(PDF-ICDD 78-2486).其中6V0M/T催化剂衍射峰强度最高, 且随着MnO2含量的增加, 催化剂的衍射图谱逐渐有所减弱, 0V6M/T催化剂最弱.这可能是因为MnOx的掺入抑制了TiO2的晶粒生长, 对于脱硝催化剂而言, 催化剂晶粒生长越完善其表面缺陷浓度相应减少, 而催化剂的晶粒生长受到抑制后表面缺陷浓度会有所提高, 这有利于催化剂脱硝活性的增强(Gao et al., 2018).另外, 在催化剂的图谱中没有观察到明显的MnOx或VOx衍射峰, 这表明MnOx和VOx物种很好地分散在二氧化钛载体中或作为无定形物种存在.
 |
| 图 3 不同催化剂样品的XRD谱 Fig. 3 XRD patterns of different catalysts |
表 1是4组催化剂的比表面积、孔容以及平均孔径数据.可以看出, 催化剂比表面积和孔容大小顺序为0V6M/T > 1V5M/T > 1V3M/T > 6V0M/T, 平均孔径大小顺序则为0V6M/T < 1V5M/T < 1V3M/T = 6V0M/T.催化剂中无定型相较多时, 介孔孔径较小但数量较多, 所以孔容和比表面积较大;而各相晶粒细化后逐渐长大后, 部分小孔塌陷, 孔径增大, 孔容和比表面积降低.因此, MnO2的掺入抑制了TiO2的晶粒生长, 有利于催化剂表面缺陷浓度的提高和比表面积的增大.比表面积增大后, 催化剂更容易吸附反应气体分子, 有利于催化剂低温活性的提高(Gao et al., 2018).
| 表 1 催化剂的比表面积、孔容和平均孔径 Table 1 Textual characteristics of different catalysts |
SCR反应活性与催化剂表面的氧化还原性能密切相关, 为了考察催化剂的氧化还原性能与活性之间的关系.选择了3组催化剂进行H2-TPR测试, 图 4给出的是0V6M/T、1V3M/T和6V0M/T催化剂的H2-TPR曲线.由图中可以看到, 3组催化剂在300~900 ℃区间内都有氢气还原, 其中400 ℃左右的还原峰可能对应着催化剂表面MnO2的还原(Zhao et al., 2017), 而500 ℃左右的还原峰对应着催化剂表面V2O5的还原(Chen et al., 2018), 更高温度的还原峰可能对应的是TiO2的还原(Xu et al., 2018).其中, 纯钒钛催化剂中掺入MnO2后低温还原峰明显增强, 即催化剂的氧化还原性能得到提高.众所周知, SCR反应中表面酸性的增强有利于催化剂高温活性的提高, 而氧化还原性能的增强有利于催化剂低温活性的增加, 这也是纯钒钛催化剂掺入MnO2后低温脱硝活性明显增强的原因之一.
 |
| 图 4 催化剂的H2-TPR曲线 Fig. 4 H2-TPR profiles of different catalysts |
X射线光电子能谱用作确定催化剂表面元素的状态, 结果如图 5所示.表 2则列出了不同催化剂表面原子浓度.如图 5a所示, Ti 2p可分为Ti3+和Ti4+两种谱峰(Yao et al., 2017).与纯钒钛或纯锰基催化剂相比, 1V3M/T催化剂Ti 2p的电子结合能向低结合能方向偏移, 即MnO2的掺入导致结合能峰的迁移, 意味着催化剂中存在电子转移.换言之, MnO2有可能和V2O5以及TiO2具有一定的强相互作用.另外, 计算了Ti3+的原子比, 如表 2所示.可以看出, MnO2掺入后Ti3+浓度有所增加, 这应该是Ti4+和Mn2+之间的电子转移使得Ti3+和Mn4+浓度增加.图 5b显示了不同催化剂的O 1s高分辨率XPS扫描图谱.O 1s峰可以拟合成两个峰, 称为化学吸附氧(以下称为Oα)和晶格氧(以下称为Oβ)(Jin et al., 2018).随着MnO2的掺入, O 1s峰显示向较高结合能偏移, 表明Mn和O原子之间存在强相互作用.据报道, 化学吸附氧是最活跃的氧物种, 能够促进NO氧化成NO2.它还可以通过“快速SCR”途径促进SCR反应的进行(Shan et al., 2018).因此, 6V0M/T、1V3M/T和0V6M/T催化剂Oα浓度分别为0.38, 0.46和0.42.可以看出, 相比6V0M/T和0V6M/T, 1V3M/T的化学吸附氧浓度最高, 即MnO2和V2O5的相互作用增加了催化剂表面化学吸附氧浓度, 有利于低温活性的提高.
 |
| 图 5 催化剂的XPS谱图 (a. Ti 2p, b.O 1s, c. V 2p, d. Mn 2p) Fig. 5 XPS profiles of different catalysts (a. Ti 2p, b. O 1s, c. V 2p, d. Mn 2p) |
| 表 2 不同催化剂表面原子浓度 Table 2 Atomic concentration of different catalysts |
图 5c显示了6V0M/T和1V3M/T催化剂V 2p的高分辨率XPS谱图.结果可分为3个谱峰:①位于517.5 eV的峰为V5+物种; ②位于516.8 eV的峰为V4+物种; ③位于516.1 eV的峰是V3+物种(Wang et al., 2018).6V0M/T的V 2p峰强度高于1V3M/T.表明催化剂表面的V2O5含量随着MnO2的掺入而降低.通过(V3++ V4+)/Vn+(n = 3、4和5)的等式计算低价钒物种的浓度比.如表 2所示, 可以看出(V3++ V4+)的比例明显降低, MnO2的掺入会导致低价钒物种向高价钒物种转化.换句话说, 这种转化增加了催化剂表面上化学吸附氧物种的相对含量.图/5d为0V6M/T和1V3M/T催化剂Mn 2p的高分辨率XPS图谱.如图 5d所示, Mn 2p谱图中, 653.6 eV位置的峰代表Mn 2p1/2(Pan et al., 2018), 而641.7 eV位置的峰则代表Mn 2p3/2(Yang et al., 2016).其中, Mn 2p3/2可以分为642.8 eV和641.8 eV两个峰, 分别代表Mn4+和Mn3+.根据前期的研究, MnOx的价态是影响其SCR活性重要因素, 不同价态的MnOx其SCR活性顺序为MnO2>Mn2O3>MnO(Chen et al., 2016).从图中可以明显看出, V2O5掺入后催化剂表面Mn4+浓度增加, 主要原因可能为V2O5的掺入抑制了Mn4+向Mn3+转化, 有利于催化剂低温脱硝催化活性的提高.
3.5 反应机理分析为研究1V3M/T上的反应机理, 进行了原位红外漫反射光谱测试, 相关结果如图 6所示.图 6a记录了在250 ℃下NO + O2和预吸附的NH3物种之间反应不同时间段的原位红外漫反射光谱.通入NO后, 所有属于NH3物种的谱峰, 如部分氧化的氨物种(NxHyOz, 1330 cm-1)(Wang et al., 2019)、酰胺物种(—NH2)和氨氧化中间体(1500~1600 cm-1)(Liu et al., 2019), 随着NO + O2通入时间的增加变得越来越弱.与此同时, 由于单齿亚硝酸盐(1370 cm-1)和桥接硝酸盐(2010 cm-1)的强度逐渐增加(Li et al., 2019), 并在5 min内完全取代了NH3物种的谱峰.此外, 5 min后出现气态或弱吸附的NO(1800、1745和1450 cm-1)(Jin et al., 2019).因此, —NH2和NxHyOz均可与NO反应, 并且它们都在SCR反应中起重要作用.其中, 酰胺类(—NH2)和氨氧化中间体对促进1V3M/T催化剂在低温下的反应起主要作用.谱图的变化表明NO + O2与预吸附的NH3物种之间的反应符合Langmuir-Hinshelwood(L-H)机理(Yang et al., 2015; Yang et al., 2017; Yang et al., 2019).
 |
| 图 6 1V3M/T催化剂的原位红外漫反射谱图 (a. NO+O2和预吸附NH3反应;b.NH3和预吸附NO+O2反应) In situ DRIFT spectra of (a. NO+O2 reacted with pre-adsorbed NH3, b. NH3 reacted with pre-adsorbed NO+O2 at 250 ℃ over 1V3M/T catalyst) |
图 6b记录了NH3和预吸附的NO + O2物种在250 ℃下不同时间段内反应的原位红外漫反射光谱.对于1V3M/T催化剂, NO + O2共吸附导致出现桥接硝酸盐(2010 cm-1)(Yuan et al., 2018), 气态或弱吸附的NO(1800、1745和1450 cm-1)(Zha et al., 2017), 双齿硝酸盐(1530 cm-1)和单齿亚硝酸盐(1370 cm-1)(Qiu et al., 2015).在通入NH3之后, 由于单齿亚硝酸盐引起的谱峰在1 min内消失, 而对应于部分氧化的氨物种谱峰产生.因此, 1300~1400 cm-1处的谱峰强度没有变化, 但峰值的中心发生了变化.对应于二齿硝酸盐和—NH2的谱峰之间存在重叠, 因此谱峰的强度(1500~1600 cm-1)没有大的变化.此外, 桥接硝酸盐(2010 cm-1)和气态或弱吸附的NO(1800、1745和1450 cm-1)不与吸附的NH3物种反应, 因此强度没有变化.换句话说, 桥接硝酸盐可以在250 ℃下稳定地存在于1V3M/T催化剂表面上.同时, 归因于吸附的NH3物种(配位NH3, NxHyOz和—NH2)的谱峰强度逐渐增加.因此, 单齿亚硝酸盐和二齿硝酸盐与1V3M/T催化剂表面上吸附的NH3物种反应, 反应速率非常高.谱图的变化表明, NH3与预吸附的NO + O2物种之间的反应也与Langmuir-Hinshelwood(L-H)机理一致, 单齿亚硝酸盐和二齿硝酸盐是主要的反应中间体.
根据上述讨论, 1V3M/T催化剂上的NH3-SCR遵循L-H机理.原位红外漫反射光谱已经证明, 在1V3M/T催化剂表面上存在吸附的单齿亚硝酸盐和硝酸齿的双齿硝酸盐与吸附的酰胺物质(—NH2)的反应.因此, 提出了1V3M/T的反应机理, 并在等式(3)~等式(8)中显示.

|
(3) |

|
(4) |

|
(5) |

|
(6) |

|
(7) |
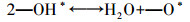
|
(8) |
1) 掺入MnOx可显著促进VOx/TiO2催化剂的脱硝活性区间由300~400 ℃向低温偏移至200~300 ℃.当MnOx和VOx物质的量的比为3 : 1时, 催化剂在250 ℃协同脱硝和脱除二英的效果最好, 催化NO和邻二氯苯效率分别为92.0%和89.0%.
2) MnOx的掺入可抑制TiO2的晶粒生长, 促进催化剂表面缺陷浓度的生成, 提高催化剂的氧化还原性能.
3) 原位红外漫反射光谱研究表明, 1V3M/T催化剂表面的脱硝反应在250 ℃时遵循L-H机理, —NH2、NxHyOz、单齿亚硝酸盐和二齿硝酸盐是主要的反应中间体.
Chen H F, Xia Y, Fang R Y, et al. 2018. The effects of tungsten and hydrothermal aging in promoting NH3-SCR activity on V2O5/WO3-TiO2 catalysts[J]. Applied Surface Science, 459: 639-646. DOI:10.1016/j.apsusc.2018.08.046 |
Chen L, Yao X J, Cao J, et al. 2019. Effect of Ti4+ and Sn4+ co-incorporation on the catalytic performance of CeO2-MnOx catalyst for low temperature NH3-SCR[J]. Applied Surface Science, 476: 283-292. DOI:10.1016/j.apsusc.2019.01.095 |
Chen Q, Guo R, Wang Q, et al. 2016. The catalytic performance of Mn/TiWOx catalyst for selective catalytic reduction of NOx with NH3[J]. Fuel, 181: 852-858. DOI:10.1016/j.fuel.2016.05.045 |
Cui Y Y, Yang G H, Xiao G H, et al. 2017. Adsorption of dioxin by bag filter plus powdered activated carbon[J]. Water Air and Soil Pollution, 228: 160-168. DOI:10.1007/s11270-017-3337-1 |
付建平, 青宪, 冯桂贤, 等. 2017. 基于污泥掺烧的某生活垃圾焚烧厂烟道气、飞灰及炉渣中的二英特征[J]. 环境科学学报, 37(12): 4677-4684. |
Gan L N, Li K Z, Xiong S C, et al. 2018. MnOx-CeO2 catalysts for effective NOx reduction in the presence of chlorobenzene[J]. Catalysis Communications, 117: 1-4. DOI:10.1016/j.catcom.2018.08.008 |
Gao C, Shi J W, Fan Z Y, et al. 2018. "Fast SCR" reaction over Sm-modified MnOx-TiO2 for promoting reduction of NOx with NH3[J]. Applied Catalysis A:General, 564: 102-112. DOI:10.1016/j.apcata.2018.07.017 |
Jin Q J, Shen Y S, Ma L, et al. 2019. Novel TiO2 catalyst carriers with high thermostability for selective catalytic reduction of NO by NH3[J]. Catalysis Today, 327: 279-287. DOI:10.1016/j.cattod.2018.04.038 |
Jin Q J, Shen Y S, Sui G R, et al. 2018. Synergistic catalytic removals of NO, CO and HC over CeO2 modified Mn-Mo-W-Ox/TiO2-SiO2 catalyst[J]. Journal of Rare Earths, 36: 148-155. DOI:10.1016/j.jre.2017.07.014 |
Li G L, Wang S X, Wu Q R, et al. 2019. Exploration of reaction mechanism between acid gases and elemental mercury on the CeO2-WO3/TiO2 catalyst via in situ DRIFTS[J]. Fuel, 239: 162-172. DOI:10.1016/j.fuel.2018.10.142 |
李雁, 郭昌胜, 侯嵩, 等. 2019. 固体废物焚烧过程中二英的排放和生成机理研究进展[J]. 环境化学, 38(4): 746-759. |
Liu H, Fan Z X, Sun C Z, et al. 2019. Improved activity and significant SO2 tolerance of samarium modified CeO2-TiO2 catalyst for NO selective catalytic reduction with NH3[J]. Applied Catalysis B:Environmental, 244: 671-683. DOI:10.1016/j.apcatb.2018.12.001 |
Liu X L, Wang J, Wang X, et al. 2015. Simultaneous removal of PCDD/Fs and NOx from the flue gas of a municipal solid waste incinerator with a pilot plant[J]. Chemosphere, 133: 90-96. DOI:10.1016/j.chemosphere.2015.04.009 |
Ning P, Song Z, Li H, et al. 2015. Selective catalytic reduction of NO with NH3 over CeO2-ZrO2-WO3 catalysts prepared by different methods[J]. Applied Surface Science, 332: 130-137. DOI:10.1016/j.apsusc.2015.01.118 |
Niu J F, Dai Y R, Yin L F, et al. 2015. Photocatalytic reduction of triclosan on Au-Cu2O nanowire arrays as plasmonic photocatalysts under visible light irradiation[J]. Physical Chemistry Chemical Physics, 17: 17421-17428. DOI:10.1039/C5CP02244D |
Pan Y, Shen Y, Jin Q, et al. 2018. Promotional effect of Ba additives on MnCeOx/TiO2 catalysts for NH3-SCR of NO at low temperature[J]. Journal of Materials Research, 33: 2414-2422. DOI:10.1557/jmr.2018.179 |
Parizek T, Bebar L, Stehlik P. 2008. Persistent pollutants emission abatement in waste-to-energy systems[J]. Clean Technologies and Environmental Policy, 10: 147-153. DOI:10.1007/s10098-007-0135-2 |
Qiu L, Pang D, Zhang C, et al. 2015. In situ IR studies of Co and Ce doped Mn/TiO2 catalyst for low-temperature selective catalytic reduction of NO with NH3[J]. Applied Surface Science, 357: 189-196. DOI:10.1016/j.apsusc.2015.08.259 |
Shan W P, Geng Y, Zhang Y, et al. 2018. A CeO2/ZrO2-TiO2 catalyst for the selective catalytic reduction of NOx with NH3[J]. Catalysts, 8: 592-604. DOI:10.3390/catal8120592 |
Wang R X, Zhang D J, Liu C B. 2017. DFT study of the adsorption of 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin on pristine and Ni-doped boron nitride nanotubes[J]. Chemosphere, 168: 18-24. DOI:10.1016/j.chemosphere.2016.10.050 |
Wang X M, Du X S, Zhang L, et al. 2018. Promotion of NH4HSO4 decomposition in NO/NO2 contained atmosphere at low temperature over V2O5-WO3/TiO2 catalyst for NO reduction[J]. Applied Catalysis A:General, 559: 112-121. DOI:10.1016/j.apcata.2018.04.025 |
Wang X X, Cong Q L, Chen L, et al. 2019. The alkali resistance of CuNbTi catalyst for selective reduction of NO by NH3:A comparative investigation with VWTi catalyst[J]. Applied Catalysis B:Environmental, 246: 166-179. DOI:10.1016/j.apcatb.2019.01.049 |
Xu L W, Wang C Z, Chang H Z, et al. 2018. New insight into SO2 poisoning and regeneration of CeO2-WO3/TiO2 and V2O5-WO3/TiO2 catalysts for low-temperature NH3-SCR[J]. Environmental Science & Technology, 52: 7064-7071. |
Xu Y F, Wu X D, Lin Q W, et al. 2019. SO2 promoted V2O5-MoO3/TiO2 catalyst for NH3-SCR of NOx at low temperatures[J]. Applied Catalysis A:General, 570: 42-50. DOI:10.1016/j.apcata.2018.10.040 |
Yang B, Huang Q, Chen MD, et al. 2019. Mn-Ce-Nb-Ox/P84 catalytic filters prepared by a novel method for simultaneous removal of particulates and NO[J]. Journal of Rare Earths, 37: 273-281. DOI:10.1016/j.jre.2018.05.023 |
Yang B, Shen Y S, Su Y, et al. 2017. Functional-membrane coated Mn-La-Ce-Ni-Ox catalysts for selective catalytic reduction NO by NH3 at low-temperature[J]. Catalysis Communications, 94: 47-51. DOI:10.1016/j.catcom.2017.02.016 |
Yang B, Zheng D H, Shen Y S, et al. 2015. Influencing factors on low-temperature deNOx performance of Mn-La-Ce-Ni-Ox/PPS catalytic filters applied for cement kiln[J]. Journal of Industrial and Engineering Chemistry, 24: 148-152. DOI:10.1016/j.jiec.2014.09.022 |
Yang N Z, Guo R T, Wang Q S, et al. 2016. Deactivation of Mn/TiO2 catalyst for NH3-SCR reaction:effect of phosphorous[J]. RSC Advances, 6: 11226-11232. DOI:10.1039/C5RA27713B |
Yao X J, Zhao R D, Chen L, et al. 2017. Selective catalytic reduction of NOx by NH3 over CeO2 supported on TiO2:Comparison of anatase, brookite, and rutile[J]. Applied Catalysis B:Environmental, 208: 82-93. DOI:10.1016/j.apcatb.2017.02.060 |
Yoshida K, Yamamoto T, Kuroki T, et al. 2009. Pilot-scale experiment for simultaneous dioxin and NOx removal from garbage incinerator emissions using the pulse corona induced plasma chemical process[J]. Plasma Chemistry and Plasma Processing, 29: 373-386. DOI:10.1007/s11090-009-9184-0 |
Yuan H Y, Sun N N, Chen J F, et al. 2018. Insight into the NH3-assisted selective catalytic reduction of NO on beta-MnO2(110):Reaction mechanism, activity descriptor, and evolution from a pristine state to a steady state[J]. ACS Catalysis, 8: 9269-9279. DOI:10.1021/acscatal.8b02114 |
Zha K, Cai S, Hu H, et al. 2017. In situ DRIFTs investigation of promotional effects of rungsten on MnOx-CeO2/meso-TiO2 catalysts for NOx reduction[J]. Journal of Physical Chemistry C, 121: 25243-25254. DOI:10.1021/acs.jpcc.7b08600 |
Zhao B H, Ran R, Guo X G, et al. 2017. Nb-modified Mn/Ce/Ti catalyst for the selective catalytic reduction of NO with NH3 at low temperature[J]. Applied Catalysis A:General, 545: 64-71. DOI:10.1016/j.apcata.2017.07.024 |
Zhao X, Huang L, Li H, et al. 2015. Highly dispersed V2O5/TiO2 modified with transition metals (Cu, Fe, Mn, Co) as efficient catalysts for the selective reduction of NO with NH3[J]. Chinese Journal of Catalysis, 36: 1886-1899. DOI:10.1016/S1872-2067(15)60958-5 |