 2020, Vol. 40
2020, Vol. 40



2. 温州市人民医院, 药学部, 温州 325000
2. Department of Pharmacy, Wenzhou People's Hospital, Wenzhou 325000
随着环保意识的日益增强, 人们对绿色可持续技术的发展愈加重视.因此, 学术界乃至整个人类社会都希望能够在社会、经济和环境之间相互制衡的基础上, 力求探索更加安全、节能的生产过程, 以获得并使用更高效和环境友好的化学产品.离子液体(ionic liquids, ILs)是由大体积有机阳离子与无机或有机阴离子组成, 熔点低于100 ℃, 在室温或室温附近温度下呈液态的盐, 具有可忽略的蒸气压、低挥发性、不易燃性、高热稳定性、化学稳定性和电化学稳定性, 尤其是其良好的可回收性, 常作为不同工艺和技术中潜在的有机溶剂替代品而备受关注(Bubalo et al., 2017;王引航等, 2018;刘宝友等, 2018; Bystrzanowska et al., 2019).然而, 离子液体本质上并非完全无毒无公害, 与常规的挥发性有机溶剂相比, 离子液体的低蒸气压虽然能够减少其对空气的污染, 但并不足以说明其暴露于空气中就是一个“绿色”的过程.工业生产过程中, 离子液体泄漏到水生环境中时, 由于其溶解度较高而导致水或土壤的污染, 同时其高稳定性又导致在环境中蓄积而成为持久性污染物, 如溴化1-烷基-3-甲基咪唑类离子液体(1-alkyl-3-methylimidazolium bromide, [Cnmim]Br)是工业界最常用的ILs之一, 已有大量研究表明这类甲基咪唑盐对水生和陆生环境中的不同生物均有一定的毒性作用, 甚至可能高于传统有机溶剂的毒性(董莹等, 2015; Wang et al., 2015).
Wang等(2014)通过实验和量子化学计算比较8种溴化咪唑类离子液体对淡水鱼的抗氧化程度, 结果表明, 这些ILs短期内即可导致鱼肝脏的氧化应激, 结合密度泛函理论(density functional theory, DFT)计算结果, 分子极化率、体积与毒性呈正相关, 而总能量与毒性呈负相关, 由此为离子液体在水生生物中的代谢途径研究提供帮助.该课题组(Wang et al., 2015)还基于24种溴化类ILs (Br-ILs)对Vibrio fischeri (V. fischeri)和Daphnia magna (D. magna)两种水生生物的毒性分别建立离子液体结构与其毒性之间的构效关系(quantitative structure-activity relationships, QSAR)模型, 结果表明, 阴离子同为溴离子的前提下, 具有不同烷基链长阳离子的离子液体在不同动物模型中表现出的毒性强弱不同, 其中, Br-ILs对V. fischeri的毒性与最低未占分子轨道能量呈负相关, 与其阳离子体积大小呈正相关; 对于D. magna动物模型, Br-ILs毒性与分子偶极矩正相关而与分子总能量负相关.为了提供更可靠的毒性评估, Cao等(2018)基于已建立的ILs毒性数据库测定119种ILs对白血病大鼠细胞系的毒性, 结果证明, 离子液体的毒性取决于亲脂性大小或者阳离子侧链长度.
ILs的阴阳离子组合数约有1018种, 因此, 常以“designer solvent”著称, 这自然与其灵活的结构可变性有关(Dean et al., 2010), 即通过将目标官能团引入阴离子或阳离子中, 可以获得具有独特的理化性质, 甚至产生不同生物活性的离子液体, 这在某种程度上很可能降低ILs的毒性, 甚至在药物开发中发挥的潜力巨大(曹建平等, 2019), 比如咪唑阳离子烷基侧链的氧化导致含水杨酸盐的ILs对细胞培养物和盐水虾卤虫的毒性显著降低(Vraneš et al., 2016);亲水性ILs进入生物体内, 与脂肪酶相互作用使后者活性受到影响而产生一系列病理变化, 而ILs浓度、阳离子烷基链长度以及阴离子形成氢键的能力都会影响酶活性(Fan et al., 2016).因此, ILs的毒性与阴、阳离子的结构和性质密切相关; 然而, 对于ILs毒性机制仍亟待进一步探索.
离子液体的种类繁多, 其微观结构和空间特征的实验研究又相当复杂而难以进行.因此, 现有的关于离子液体作用的分子机制研究大多是通过理论方法获得的. Wang等(2011)将芘作为荧光探针, 对水溶液中[Cnmim]Br (n=4、6、8、10、12)和[C4mim][BF4]分别与小牛胸腺(calf thymus DNA-D1501, ctDNA)相互作用进行动力学模拟, 结果表明, ILs-ctDNA的结合吉布斯能与结合常数随着烷基阳离子的链长增加而增加, 其中, DNA与阳离子头基之间的静电相互作用是主要驱动力, 而与烷基烃链之间的疏水作用同样不可忽略. Ding等(2010)通过多种谱学方法对水相中的[C4mim]Cl与DNA之间的相互作用进行表征, 发现二者结合过程中DNA仍保持正常的生理构象, 但碱基之间的堆积作用和螺旋结构发生一定程度的改变, 一旦改变IL与DNA的物质的量的比, IL在DNA表面上独特的结合模式导致形成具有不同结构的DNA-IL复合物.阳离子除了与DNA磷酸骨架以静电结合, 与大沟发生静电和范德华相互作用之外, 更重要的是, 无论阴离子类型如何, 阳离子还与DNA小沟中富含AT碱基对序列优先结合(Chandran et al., 2012; Nakano et al., 2014).
本项工作旨在通过量子化学方法计算探索离子液体与碱基之间的相互作用模式、能量学和电子结构特征.采用DFT方法计算并分析水环境中阳离子([Cnmim]+)、阴离子(Br-)、离子对([Cnmim]Br, n=2、4、6、8、10)与胸腺嘧啶(thymine, T)相互作用体系, 在改变碱基与离子液体的化学计量比的过程中形成一系列稳定结构, 以深入探讨二者之间相互作用的本质.
2 计算方法(Computational methods)M06-2X交换相关泛函描述生物体系之间的非共价相互作用具有一定的优势, 如DNA体系中碱基之间的氢键和堆积作用(Gu et al., 2011), 两性离子簇之间的色散和离子型氢键相互作用(Walker et al., 2013), 而本课题组前期研究工作(Xu et al., 2018;徐佳禛等, 2018)同样证明了该泛函描述非共价相互作用时表现出的良好性能.为进一步深入研究碱基-离子液体作用体系(thymine-ILs, T-ILs), 本文运用M06-2X/6-311++G(2d, p)方法, 采用极化连续介质模型(polarizable continuum model, PCM)模拟水溶液(介电常数ε=78.4), 计算胸腺嘧啶分别与咪唑阳离子([Cnmim]+)、溴阴离子(Br-)和离子对([Cnmim]Br, n=2、4、6、8、10)作用体系的结构和能量学特征, 由此分析碱基与离子(对)之间的作用模式和作用强度以及阳离子烷基链长对体系稳定性的影响; 基于每种模式下的最稳定体系, 将胸腺嘧啶与离子液体的物质的量的比由1 : 1增至1 : 2, 即增加离子液体数目使碱基主要受体和供体位点得到饱和; 对逐步增加的离子簇体系展开分子中原子(atoms in molecule, AIM) (Popelier, 2000)之间的拓扑结构, 自然键轨道(natural bond orbital, NBO) (Kasende et al., 2016)电子结构分析.所有计算均由Gaussian 16程序包(Frisch et al., 2016)完成, 结合Multiwfn程序, 通过AIM理论方法量化离子液体与碱基之间的相互作用强度.所有优化后的结构能量均采用Boys和Bernard(2006)的Counterpoise(CP)方法进行基组重叠误差(basis set superposition errors, BSSE)校正.采用超分子方法(the supermolecule approach) (Chałasiński et al., 2000;Tateishi-Karimata et al., 2018)计算相互作用能(Eint), 表征复合物与构成该复合物各碎片之间的能量差异, Eint数值大小用于衡量分子间相互作用强度.即:
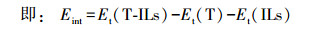
|
(1) |
式中, ILs离子([Cnmim]+或Br-)或离子对([Cnmim]Br), Et为考虑零点能(zero-point vibrational energy, ZPVE)校正后的电子能量.
形成复合物的过程中, 各碎片的几何结构发生一定程度的变化, 这一能量差异称为变形能Edef, 即碎片在独立状态下, 由复合物中的碎片单点能量减去单体状态下的能量, 其数值总为正值.这里本课题组主要关注胸腺嘧啶的变形能(EdefT).即:
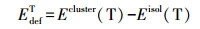
|
(2) |
体系结合能(Ebind)来自相互作用能与变形能之和(Eint + Edef), 用于表征体系的稳定性. Ebind数值愈负, 表明该复合物更稳定, 也就是分子间相互作用在能量上愈有利.
3 结果和讨论(Results and discussion) 3.1 碱基和离子(对)胸腺嘧啶存在6种可能的互变异构体, 所有的实验结果均与二酮形式的异构体一致, 后者在凝聚相和气相中都是最稳定的形式(Kasende et al., 2016).因此, 本研究对二酮式胸腺嘧啶分别与阳离子、阴离子和离子对的相互作用体系进行分析和讨论.图 1为优化后的胸腺嘧啶和咪唑阳离子的分子表面静电势(molecular electrostatic potential, MEP)分布图, 这将有助于从定性层面预测碱基与离子之间发生相互作用的可能位点, 其中黄色小球对应静电势极大点, 青色小球对应极小点, 静电势极大值和极小值以及分子中主要的原子编号已在图中标识.
在前期研究工作中(Xu et al., 2018), 本课题组已在4组计算水平(M06-2X/6-311++G(2d, p)/gas; M06-2X/6-311++G(2d, p)/PCM/water; ωB97XD/6-311++G(2d, p)/gas; ωB97XD/6-311++G(2d, p)/PCM/water)下对[Cnmim]Br离子对进行优化并获得共32种稳定结构.其中, 采用M06-2X/6-311++G(2d, p)/PCM/water方法优化的结构能量学参数值已给出.
 |
| 图 1 胸腺嘧啶(T)和咪唑阳离子([Cnmim]+)的3D结构及其静电势等值面图 Fig. 1 Schematic structures and molecular electrostatic potential maps of thymine (T), 1-alkyl-3-methylimidazole cation ([Cnmim]+) |
如图 1所示, 胸腺嘧啶具有2个顺式酰胺基团(N1—H和N3—H, 位于最正的红色静电势区)和2个羰基(C2=O7和C4=O8, 位于最负的蓝色静电势区), 可分别作为良好的氢键供体和受体参与非共价作用的形成.咪唑阳离子头基(C2/4/5—H)的静电势呈红色, 尤其是C2—H附近的静电势数值最高, 随烷基侧链长度的增加呈递减趋势.考虑到带电体系与碱基π电子云之间的堆积相互作用对于识别核苷酸和核酸的重要性(Hong et al., 2001; Fiethen et al., 2008;Kataev et al., 2016), 碱基与离子(对)之间的堆积作用同样不可忽视.
3.2.1 T-[Cnmim]+体系本课题组在M06-2X/6-311++G(2d, p)/PCM/water理论水平下对胸腺嘧啶分别与[C2mim]+、[C4mim]+、[C6mim]+、[C8mim]+和[C10mim]+咪唑阳离子作用体系进行优化计算, 各自得到11种稳定结构.根据咪唑环与胸腺嘧啶的相对位置, 可将T-[Cnmim]+体系划分为堆积(stacking, S)、垂直(perpendicular, P)和共平面氢键(coplanar HB, H)3种构型, 而由阳离子作用碱基的位点不同又可细分为S1(C2=O7位点)和S2(C4=O8位点), P1和P2, H1和H2.
(1) T-[Cnmim]+堆积构型堆积构型中, 嘧啶环与咪唑环彼此平行, 阳离子烷基C6—H键垂直于咪唑环并与羰基氧原子形成C—H…O相互作用.如表 1所示, 对于给定的侧链碳数, 阳离子作用于碱基同一受体位点的能量学参数值相差并不大. Ebind数值结果显示, 含不同烷基侧链的阳离子作用于碱基的不同位点时, 所有结构的稳定性顺序一致, 即T-[Cnmim]+: 1>2>3>4.相对于C4=O8位点, 阳离子更倾向于靠近C2=O7.此外, C6—H…O7中的氢原子和氧原子之间距离范围在2.483~2.519 Å之间变化, 数值均小于氢原子和氧原子之间的范德华半径总和(2.72 Å) (Petti et al., 1988), 其键角在134.79°~135.15°范围内, 数值均超过110° (Ricca et al., 1995), 表明T-[Cnmim]+体系的堆积构型中, 碱基与阳离子之间还有氢键相互作用的形成, 结合C2=O7和C6—H基团几乎可忽略的键长变化(约伸长0.001 Å), 可知该氢键作用强度较弱.最稳定的T-[Cnmim]+-1复合物中, π…π+之间的距离介于3.383~3.420 Å之间, 胸腺嘧啶和阳离子的几何结构与各自的单体相比无明显变化, 这种可忽略的变化趋势并不依赖于阳离子烷基链长度, 较小的EdefT数值同样体现出复合物中碱基的稳定性.仔细比较T-[Cnmim]+-1体系中C6—H…O7之间的氢键距离及键角大小, 发现T-[C10mim]+-1结构中C6—H…O7氢键作用最强, 相应的距离(2.483 Å)最短、角度(135.15°)最大, 但Eint绝对值却体现了所谓的“侧链效应”(Pham et al., 2010), 即分子间相互作用强度随烷基链长增加而增强, 但n>8之后出现“截止效应”(Wu et al., 2018), 强度不再增加(16.56 < 16.61 < 16.80 < 17.86>17.15 kJ · mol-1), 表明C—H…O氢键相互作用在堆积结构中能够提供额外的稳定性, 但并非主要的稳定作用力.
| 表 1 单个咪唑阳离子作用于胸腺嘧啶(T-[Cnmim]+)的堆积构型对应的结合能、相互作用能、碱基变形能和BSSE校正能 Table 1 Calculated energetic characteristics of the mono-cationized thymine for the stacking T-[Cnmim]+ complexes including BSSE correction |
(2) T-[Cnmim]+垂直构型阳离子与嘧啶环呈T型, 其头基C2—H和烷基C6—H通过氢键与碱基中的羰基氧原子形成弱相互作用.比较Ebind数值, 两种垂直构型的稳定性顺序如下:
T-[C2/6mim]+: 5>6>7>8
T-[C4/8mim]+: 5>7>6>8
T-[C10mim]+: 5>8>6>7
不同阳离子体系之间, 各结构稳定性顺序稍有不同, 但最稳定的结构一致为T-[Cnmim]+-5 (图 2), 即碱基中的C4=O8基团与阳离子头基和C6—H分别形成1根氢键, 相应的距离和氢键键角分别在2.113~2.171 Å, 142.88~148.84° (C2—H…O8)和2.403~2.455 Å, 148.39~149.36° (C6—H…O8)范围内, C4=O8键大约伸长0.005 Å, C2—H和C6—H键分别伸长0.002 Å和0.001 Å, 表明这两根氢键各自在不同阳离子-碱基体系中的作用强度差异不大, 其中咪唑头基C2—H的氢键供体能力比烷基C6—H强得多, 这是由于咪唑环中的C=N π+键缺电子使C2—H氢原子酸性最强(Araujo et al., 2011; Bubalo et al., 2014).进一步比较T-[Cnmim]+-5的Eint绝对值, 较长烷基链(n ≥ 4)中同样出现“侧链效应”, n>8后出现“截止效应”(12.63 < 13.33 < 14.06>12.43 kJ · mol-1).
 |
| 图 2 T-[Cnmim]+ (n=2、4、6、8、10)体系堆积(S), 垂直(P)和共面(H)构型分别对应的最稳定结构 Fig. 2 Geometries of the most stable stacking, perpendicular, and coplanar T-[Cnmim]+ (n=2, 4, 6, 8, 10) complexes |
(3) T-[Cnmim]+共面构型对于给定的阳离子, 胸腺嘧啶碱基中的2个羰基氧原子与咪唑离子作用体系的稳定性如下:
T-[C2/4mim]+: 9>10>11
T-[C6/8/10mim]+: 9>11>10
碱基分别与较短烷基链(n=2、4)和与较长烷基链(n=6、8、10)阳离子作用时稳定性高低顺序也稍有不同, 但最稳定的结构一致为T-[Cnmim]+-9 (图 2), 与垂直构型一样, 碱基的C4=O8基团分别与阳离子的C2—H和C6—H基团形成2根氢键, 相应的距离及键角分别在2.113~2.120 Å, 145.58~146.17° (C2—H…O8)和2.357~2.369 Å, 150.04~150.30° (C6—H…O8)范围内, 与单体状态下的碱基相比, C4=O8键约伸长0.004 Å, 与单体状态下的阳离子相比, C2—H和C6—H键长几乎不变.比较T-[Cnmim]+-9对应的Eint绝对值, 较短和较长烷基链作用体系同样存在“侧链效应”, 而后者n>8之后也出现“截止效应”(12.23 < 12.86 kJ · mol-1; 12.45 < 13.01>11.31 kJ · mol-1).
图 3直观比较了T-[Cnmim]+-1、T-[Cnmim]+-5、T-[Cnmim]+-9复合物中的结合能和分子间相互作用能量数值, 伴随有氢键的堆积结构都是最稳定的, 分子间相互作用强度显著强于垂直和共面结构.这种π-π+作用存在于许多生物大分子或金属-有机物体系中, 例如带正电荷的组氨酸残基与酪氨酸、苯丙氨酸等中性芳香族氨基酸之间的相互作用在稳定蛋白质结构和配体结合过程中起重要作用(Cauët et al., 2005), 含氮芳香阳离子喹啉和中性π体系之间存在强烈吸引力, 甚至比π-π作用更强(Karimi et al., 2019).因此, T-[Cnmim]+体系中, 分子间相互作用以π-π+堆积为主, C—H…O氢键作用为辅.
 |
| 图 3 最稳定的T-[Cnmim]+-1、T-[Cnmim]+-5、T-[Cnmim]+-9结构对应的结合能(实线)与相互作用能(虚线) Fig. 3 The binding (filled symbols) and interaction energies (empty symbols) for T-[Cnmim]+ complexes based on the most stable stacking, perpendicular, and coplanar configurations (S, P, H) |
 |
| 图 4 T-[Cnmim]Br (n=2、4、6、8、10)体系堆积、垂直和共面3种构型对应的最稳定结构 Fig. 4 Geometries of the most stable stacking, perpendicular and coplanar T-[Cnmim]Br (n=2, 4, 6, 8, 10) complexes |
 |
| 图 5 最稳定的T-[Cnmim]Br-1、T-[Cnmim]Br-5、T-[Cnmim]Br-9结构对应的结合能(实线)与相互作用能(虚线) Fig. 5 The binding (filled symbols) and interaction energies (empty symbols) for T-[Cnmim]Br complexes based on the most stable stacking, perpendicular, and coplanar configurations (FS, FP, FH) |
 |
| 图 6 M06-2X/6-311++G(2d, p)/ PCM/water理论水平下优化得到的T-2[Cnmim]Br (n=2、4、6、8、10)体系中最稳定的堆积结构 Fig. 6 Optimized geometries for the full-ionized thymine molecule interacting with ion cluster (T-2[Cnmim]Br, n=2, 4, 6, 8, 10) at the M06-2X/6-311++G(2d, p)/ PCM/water level of theory |
根据静电势图可初步判断, 溴离子位于嘧啶环N1—H和N3—H附近并与之形成非共价作用的可能性较大, 考虑到阴离子-π相互作用, 本研究在同一理论水平下计算得到4种结构.根据溴离子相对于嘧啶环平面的位置分为面内(front, T-Br--1, T-Br--2)和面外(top, T-Br--3, T-Br--4)两种(Hunt et al., 2015).在2种front构型中, 阴离子与碱基均形成单根氢键, H…Br距离分别为2.349、2.417 Å, 相应的氢键键角分别为160.89° (N1—H…Br), 179.44° (N3—H…Br), 相较于碱基单体, N1—H和N3—H键分别伸长约0.015、0.014 Å.
比较Ebind数值, T-Br--1相对更稳定, 正如文献(Araujo et al., 2011)中醋酸根阴离子更倾向与尿嘧啶中的N1—H酰胺基团形成氢键.而对于平面外构型, 溴离子无论位于嘧啶环上方还是下方, π…Br作用相对于N—H…Br氢键作用极其不稳定, 前者相互作用能量也明显更高, 表明碱基与溴离子之间以N—H…Br氢键相互作用占主导.
| 表 2 单个溴阴离子作用于胸腺嘧啶碱基(T-Br-)的两种构型对应的结合能、相互作用能、碱基变形能和BSSE校正能 Table 2 Calculated energetic characteristics of the mono-anionized thymine for the front and top T-Br- complexes including BSSE correction |
为考察阴阳离子同时作用于碱基的情况, 基于同一理论水平, 本课题组对于给定的阳离子侧链碳数n, 优化得到了12种稳定的T-[Cnmim]Br结构.同样地, 胸腺嘧啶与[Cnmim]Br离子对之间存在3种不同的构型:面对面堆积(face-to-face stacking, FS)、边对面垂直(edge-to-face perpendicular, FP)和共面氢键(face-to-face coplanar, FH).
(1) T-[Cnmim]Br堆积构型对于堆积构型, 阳离子以错位平行的方式在嘧啶环上方或下方通过氢键作用于羰基氧原子, 而阴离子与碱基和阳离子同时形成氢键相互作用. Ebind数值显示4种结构的稳定性顺序为:
T-[C2/46/8mim]Br: 1>2>3>4
T-[C10mim]Br: 1>2>4>3
最稳定的结构一致为T-[Cnmim]Br-1, π…π+之间的距离为3.395~3.449 Å, 两两之间相互作用的主要氢键长度及角度分别在2.426~2.434 Å和145.77~146.14° (N1—H…Br), 2.445~2.450 Å和131.04~131.45° (C2—H…O7), 2.827~2.835 Å, 118.01~118.44° (C2—H…Br)范围内. N1—H和C2=O7键相对于碱基单体分别伸长约0.012 Å和0.003 Å, 胸腺嘧啶与溴离子的静电吸引强于碱基与阳离子之间的氢键作用, 类似于尿嘧啶在1, 3-二烷基咪唑乙酸盐离子液体中的溶剂化过程, 碱基与阴离子之间的氢键作用是促进碱基溶解的主要驱动力, 但尿嘧啶与咪唑阳离子之间的氢键作用同样不可忽略(Araujo et al., 2011).就离子液体而言, 胸腺嘧啶的加入使C2—H和C6—H键各自缩短约0.005 Å和0.01 Å, 结合C2—H…Br之间的距离更长, 角度明显更小, 表明阴阳离子间的氢键作用减弱.中T-[Cnmim]Br-1复合物的Eint绝对值结果显示, T-[C2mim]Br-1体系作用能量最低, n ≥ 4时, 分子间相互作用强度与阳烷基链长呈“侧链效应”, n>8后又出现“截止效应”(35.69>33.80 < 34.17 < 34.62>34.19 kJ · mol-1).
(2) T-[Cnmim]Br垂直构型FP构型中, 溴离子位于咪唑环上方并通过氢键与胸腺嘧啶N—H基团形成面内相互作用, 同时, 阳离子C2—H和C6—H键以边对面的方式同时指向碱基的羰基氧并形成氢键.与T-[Cnmim]+体系一样, 由于较长烷基链存在一定的空间位阻, 胸腺嘧啶的羰基氧作为良好的氢键受体, 更偏向于靠近3位甲基质子而不是1-烷基链, 这种倾向同样发生在CO2-[C4mim]+体系中(Corvo et al., 2013);不同的是, 阳离子倾向于作用C2=O7位点. 4种结构的稳定性顺序依次为:
T-[C2mim]Br: 5>6>7>8
T-[C4/6/810mim]Br: 5>7>6>8
阴阳离子以邻位方式(T-[Cnmim]Br-5, 6, 7)作用于碱基的体系稳定性高于对位方式(T-[Cnmim]Br-8), 这两种作用方式在不同阳离子体系中的分子间相互作用强度差异高达15.20~18.49 kJ · mol-1, 表明阴阳离子之间的协同作用有助于增加体系的稳定性.
随着烷基链的延长, 碱基-离子对体系最稳定的垂直结构均为T-[Cnmim]Br-5, 阴、阳离子分别与碱基形成1根和2根氢键, 相应的长度和角度变化较小, 与独立碱基相比, N1—H和C2=O7键各自伸长0.020 Å和0.010 Å.比较T-[Cnmim]Br-5垂直结构的Eint数值, 分子间相互作用随烷基链长增加而增强, 但n>4之后出现“截止效应”(33.02 < 33.75>32.85 < 32.89 < 33.26 kJ · mol-1), 这与土壤中草地夜蛾暴露于[Cnmim]Br (n = 2、4、6、8、10)中, [C4mim]Br的细胞毒性最大是一致的(Wu et al., 2018).
(3) T-[Cnmim]Br共面构型碱基与离子对共同形成3根氢键, 相应结构的稳定性高低按如下排序:
T-[C2mim]Br: 9>10>11>12
T-[C4/6/8/10mim]Br: 9>11>10>12
与垂直构型一致, 离子对之间以邻位方式(T-[Cnmim]Br-9, 10, 11)作用于碱基的体系稳定性强于对位作用模式(T-[Cnmim]Br-12).进一步观察中邻位作用最稳定的结构(T-[Cnmim]Br-9), 这与Watson-Crick碱基对之间的“Watson-Crick”氢键作用位点(N3—H和C4=O8) (Crick et al., 1954)不同, 对于前者, 阴、阳离子分别与N1—H和C2=O7, 即所谓的“sugar edge”位点(Leontis et al., 2002)形成氢键相互作用, 长度和角度范围分别处于2.318~2.326 Å和179.33~179.91° (N1—H…Br), 2.237~2.257 Å和133.72~135.29° (C2—H…O7), 2.320~2.348 Å和146.39~147.75° (C6—H…O7)范围内, N1—H和C2=O7键长变化也伸长了0.020 Å和0.010 Å, 因此FP和FH构型中, 碱基与阴、阳离子之间的氢键相互作用强度相近.此时, Eint数值结果同样显示n > 4之后出现“截止效应”.不同构型中最稳定的T-[Cnmim]Br结构绝不是简单地由最稳定单离子作用体系(T-[Cnmim]+或T-Br-)组成的. 3种构型对应的最稳定T-[Cnmim]Br-1、T-[Cnmim]Br-5和T-[Cnmim]Br-9结构中, 与最稳定的碱基水合物一样(Pullman et al., 1978), 阴、阳离子总是占据“sugar edge”位点, 与胸腺嘧啶形成N1—H…Br和C—H…O7氢键相互作用, 这两种氢键作用在FS构型中明显弱于另外两种构型.然而, 结合能量学参数来看, 尽管堆积构型中的BSSE校正能相对更高, 导致Eint和Ebind绝对值略有降低, 但校正前后分子间相互作用强弱顺序仍一致为:面对面堆积>边对面垂直>共面氢键, 表明T-[Cnmim]Br复合物中π…π+作用强于碱基与阳离子之间的氢键作用, 前者与N1—H…Br氢键相互作用的协同性更强, 这种协同性在碱金属与碱土金属离子之间同样存在(Vijay et al., 2008).
基于堆积、垂直和共面3种构型对应的最稳定T-[Cnmim]+-1、T-Br-1、[Cnmim]Br-1、T-[Cnmim]Br-1体系中结构, 本课题组进一步考察碱基对离子(对)的偏好或离子(对)对碱基化学结构的影响.结果可知, 随着阳离子烷基链的延长, 任一碱基-离子(对)体系中, T、[Cnmim]+或[Cnmim]Br碎片的几何结构相对于各自单体状态下的变化不大, 因此, 同一体系中, 烷基链长对其稳定性的影响差异不大, 动力学模拟结果也显示了烷基链长对DNA稳定性影响很小(Jumbri et al., 2014).
| 表 3 单个离子对作用于胸腺嘧啶(T-[Cnmim]Br)的面对面堆积构型对应的结合能、相互作用能、碱基变形能和BSSE校正能预测值 Table 3 Calculated energetic features of the di-ionized thymine for the stacking T-[Cnmim]Br complexes including BSSE correction |
图 7直观显示了各稳定体系对应的Eint数值, 结果表明, 无论胸腺嘧啶相对于咪唑阳离子的位置如何, 分子间相互作用强度一致遵循如下顺序: T-[Cnmim]Br>T-Br->[Cnmim]Br>T-[Cnmim]+.分子间两两相互作用协同增强T-[Cnmim]Br复合物的稳定性; 阴离子对胸腺嘧啶的亲和力强于离子液体本身, 这可以解释为碱基与阴离子和阴阳离子之间的结合存在竞争性, 最终使阴阳离子之间的氢键遭到破坏, 导致阴离子与碱基形成了新的分子间相互作用(Araujo et al., 2011).
 |
| 图 7 最稳定的T-[Cnmim]+、T-Br-、[Cnmim]Br、T-[Cnmim]Br和T-2[Cnmim]Br体系中堆积, 垂直和共面结构对应的分子间相互作用强度折线图 Fig. 7 Calculated interaction energy values of the three most stable configurations for T-[Cnmim]+, T-Br-, [Cnmim]Br, T-[Cnmim]Br and T-2[Cnmim]Br complexes |
 |
| 图 8 AIM结合IGM等值面图可视化碱基-离子液体体系的弱相互作用(δginter =0.01 a.u.) Fig. 8 AIM analysis of ionized thymine complexes at critical points (BCPs for orange ball and CCPs for magenta color) by means of the visualized IGM isosurfaces (δginter=0.01 a.u., blue stands for attractive interactions and green for very weak van der Waals-type interactions) |
DNA的结构可以通过改变离子液体的浓度加以调节(Vijay et al., 2008;Ding et al., 2010).为了进一步探索碱基与离子液体之间的相互作用规律, 针对T-[Cnmim]Br体系堆积、垂直和共面3种构型对应的最稳定结构, 增加碱基与离子对之间的摩尔比, 即加入第二个离子对扩大体系, 最终优化得到每种构型对应的碱基-离子液体超分子结构.对比表 4中Ebind数值, 结果显示Br-…T…Br-氢键与π+…π…π+联合堆积作用之间的强协同作用依然导致T-2[Cnmim]Br-1结构更有利, 尽管BSSE校正能明显高于另外两种结构, 但这并不影响3种构型的相对稳定性.
| 表 4 2个离子对作用于胸腺嘧啶(T-2[Cnmim]Br)的面对面堆积构型对应的结合能、相互作用能、碱基变形能和BSSE校正能预测值 Table 4 Calculated energetic features of the full-ionized thymine for the stacking, perpendicular and coplanar T-[Cnmim]Br complexes including BSSE correction |
溴离子与胸腺嘧啶中的N1—H和N3—H质子以及阳离子头基和烷基质子形成氢键, 两个咪唑环分别位于碱基平面环上、下方, 环环之间以错位堆积呈近似平行.结果表明, 对于嘧啶环上方的离子对, π…π+堆积距离介于3.340~3.453 Å之间, 与等当量的T-[Cnmim]Br-1体系相比, C2—H…Br, N1—H…Br和C6—H…O7氢键距离变化不大, 同时C2—H、N1—H和C6—H键长几乎不变, 而C2=O7键和C4=O8键分别伸长了约0.006 Å和0.003 Å, 这是由于加入第二对离子液体(位于嘧啶环下方的离子对)后, 嘧啶环下方的阳离子C6—H仍与C2=O7氧原子形成氢键作用.溴离子与N3—H之间的氢键距离和角度介于2.396~2.402 Å和176.16°~176.88°之间, 相互作用强于N1—H…Br (距离为2.408~2.428 Å, 角度为146.09~146.68°), 碱基与下方阳离子之间的π…π+距离介于3.375~3.391 Å之间.更多氢键与堆积作用的参与最终导致更强的亲和力(图 7), 与T-[C2mim]Br-1相同, T-2[C2mim]Br-1体系中分子间相互作用最强, n>8后出现“截止效应”(71.93>68.96>68.76 < 69.62>69.10 kJ · mol-1).
3.2 AIM分析为了更准确地描述分子间相互作用类型及其强度, 特别是碱基与阳离子之间的作用, 本研究采用Bader及其同事的AIM理论研究最稳定T-[Cnmim]+-1、T-Br-1、T-[Cnmim]Br-1、T-2[Cnmim]Br-1体系的电子结构, 通过(3, -1)键临界点(bond critical point, BCP)和(3, +3)笼临界点(cage critical point, CCP)处的电子密度(ρ)及其拉普拉斯函数(▽2ρ)值符号和大小对原子之间相互作用的性质进行表征.通常ρ值用于描述键的强度, 数值越大键强度越强; ▽2ρ值用于描述键的类型. ▽2ρ>0表示闭壳层相互作用, 具有氢键、离子键、配位键或者范德华相互作用的特征(Qu et al., 2012).根据IUPAC对X—H…Y氢键的描述, H和Y原子由BCP连接(Jumbri et al., 2016), 典型的X—H…Y氢键符合ρBCP=0.002~0.035 a.u., ▽2ρBCP=0.024~0.139 a.u.标准(Koch et al., 1995;Popelier, 1998), 若ρ < 0.01 a.u., 并非表示不存在氢键, 而是表明该氢键以离子或色散成分为主(Bader et al., 1984; Parthasarathi et al., 2006; Hun, 2015), 比如C—H…π作用.此外, 对于以范德华力作用为主的复合物, 还存在CCP, 该处的电子密度ρCCP较小且与▽2ρCCP同为正值.电子密度及其拉普拉斯值的累积(∑ρBCP, ∑ρCCP和∑▽2ρBCP, ∑▽2ρCCP)用于表征π+…π相互作用(Vijay, 2010; Azizi et al., 2019).
为更加直观地可视化分子间弱相互作用, 本研究采用独立梯度模型(independent gradient model, IGM)(Lefebvre et al., 2017)考察片段间的δg实空函数, 中密集的网格即为δg=0.01 a.u.的等值面图.等值面蓝色区越大者, ρ较大, 若接近0.01 a.u., 则以静电吸引的氢键作用为主, 绿色区越大则对应较小的ρ值, 符合相互作用较弱的范德华体系, 后者本质上具有色散吸引的性质. T-[Cnmim]+-1体系中, 3~4个BCPs和2个CCPs用于表征π+…π相互作用, 1个BCP描述C6—H…O7氢键, π电子云堆积(∑ρBCP≈ 0.019~0.025 a.u.; ∑ρCCP≈ 0.010 a.u.)明显强于C6—H…O7氢键(ρBCP≈ 0.009 a.u.)相互作用, 后者电子密度小于0.01 a.u., 具有色散吸引的性质. T-Br--1中, N1—H…Br之间的ρBCP (≈ 0.023 a.u.)和▽2ρBCP (≈ 0.054 a.u.)数值均在典型的X—H…Y氢键标准范围内.阴阳离子同时作用于碱基时, 共找到8个BCPs和2个CCPs, 其中碱基-阴离子和阴阳离子之间的相互作用各由2个BCPs表征, 其余表征碱基-阳离子作用, 结果表明, 相较于T-Br--1, N1—H…Br之间的电子密度减小, 增加了C6—H…Br作用, 其ρBCP和▽2ρBCP值分别小于0.01 a.u.和0.024 a.u, 因此这种离子型氢键很弱; 与T-[Cnmim]+-1相比, C6—H…O7氢键作用稍微增强, π+…π之间的∑ρCCP数值略有减少, 但∑ρBCP数值在T-[C2/4mim]+-1体系略增加0.001 a.u., 在T-[C6/8/10mim]+-1体系却分别降低了0.01 a.u.、0.005 a.u.、0.004 a.u..值得注意的是, T-[C6mim]+-1结构中π+…π之间仅找到3个BCP, ∑ρBCP数值较低.此外, 随着烷基链的延长, 阴阳离子之间的ρBCP数值无明显差异, 这与烷基链长对体系中[Cnmim]Br碎片的几何结构影响不大的结果一致.
碱基-离子液体饱和体系中, 分子间总共找到15~16个BCPs和3个CCPs, 其中2~3个BCPs表征N—H…Br氢键, 有4个BCPs表征阴阳离子间氢键, 其余8~9个BCPs中, 2个表征C6—H…O7作用, 1个表征C7—H…π弱氢键, π+…π堆积由剩余BCPs和3个CCPs表征, 结果显示, T-2[Cnmim]Br饱和体系中所涉及的分子间相互作用除了存在C7—H…π额外的弱氢键作用, 与T-[Cnmim]Br-1体系并无差异, 但由于前者结构中更多氢键和堆积作用的形成导致片段之间两原子在临界点处的电子密度大大增加, 分子间相互作用也随之增强.
3.3 NBO分析 3.3.1 NPA电荷布居分析本课题组再次基于以上4种最稳定体系, 采用自然布居分析(natural population analysis, NPA)方法(Reed et al., 1985)分析并考察碱基与离子(对)发生相互作用后各片段(胸腺嘧啶、溴离子、咪唑阳离子)相对于相应游离状态下的电荷转移情况. T-[Cnmim]+-1体系中, 烷基链长对片段间的电荷转移影响不大, 胸腺嘧啶电荷数由0.000 a.u.增加到0.004 a.u., 咪唑阳离子由1.000 a.u.降至0.996 a.u., 因此二者结合过程中主要是由π电子云向π+正离子发生电子转移, 尽管存在C6—H…O7弱氢键相互作用, 此时胸腺嘧啶作为质子受体往往得到电子而使原子电荷有所减少. T-Br--1体系中, 胸腺嘧啶电荷数减少了0.005 a.u., 溴离子由-1.000 a.u.减少到-0.995 a.u., 表明二者结合过程中由阴离子向碱基发生电子转移.
另外, 胸腺嘧啶在阴离子体系中的电荷转移量明显高于阳离子体系, 再次证明了碱基对阴离子更高的敏感性. T-[Cnmim]Br-1体系中, NPA电荷转移量依然不依赖烷基链长度, 阴、阳离子电荷转移量分别为0.057 a.u.和0.019 a.u., 比在单个碱基-阴(阳)离子体系中的转移量多, 体现了阴阳离子同时作用于碱基的协同性, 胸腺嘧啶的NPA电荷减少, 转移量约0.038 a.u., 表明胸腺嘧啶整体上是得电子的, 碱基向π+电子云发生的电子转移不再占主导.最后, 对于T-2[Cnmim]Br饱和体系, 由于第二个离子对的加入, 随着烷基链延长, 每个片段的电荷转移量分别在0.049~0.054 a.u. (T)、0.987~0.990 a.u. ([Cnmim]+)、0.936~0.939 a.u. (Br-)、0.049~0.054 a.u.范围内变化.
3.3.2 NBO轨道分析为进一步研究逐步饱和过程中, 碱基与离子(对)之间相互作用的本质, 本研究对上述4种稳定体系展开NBO分析, 并以二阶稳定化能E2>2 kJ · mol-1作为基准(Kasende et al., 2016), 该数值越大, 表明电子供体轨道i与受体轨道j之间的相互作用越强(Glendening et al., 2013).
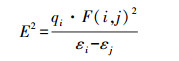
|
(3) |
式中, qi是供体占据轨道的电荷数, εi和εj为对角线元素, F(i, j)为Fock矩阵元.对于ππ+相互作用, 以∑Eπ→π+2和∑Eπ←π+2分别表示碱基向π+阳离子和π+电子云流向碱基的电荷转移能(Kasende et al. 2016; Azizi et al. 2019).如表 5所示, T-[Cnmim]+-1体系中, ππ+稳定化能明显高于O7孤电轨道与阳离子C6—H反键轨道之间的相互作用能, 而碱基作为供体轨道, 其π成键轨道与π+反键轨道之间的相互作用能又显著强于π+反键与π成键轨道间的稳定能(∑Eπ→π+2>∑Eπ←π+2), 这与胸腺嘧啶电荷数增加的结果是一致的. T-Br--1体系中, 氢键稳定化能由溴离子的孤对电子轨道与胸腺嘧啶的N1—H反键(σN1—H*)轨道之间的相互作用, 能量高达73.48 kJ · mol-1, 明显高于碱基-阳离子体系.当阴阳离子同时作用于胸腺嘧啶时, 碱基与阴、阳离子两两之间均存在相互作用, 其中, 碱基与阳离子之间的轨道相互作用能大大降低, 仅存在较弱的π→ π+*和lpO7→ σC6—H*轨道作用, 与溴离子之间的稳定化能主要源于lpBr-→ σN1—H*轨道作用, 此时, lpBr-→ σN1—H*轨道作用占绝对主导, 阴阳离子之间的轨道作用(lpBr-→ σC—H*和lpBr-→ π+*)甚至强于碱基与阳离子作用, 从而提供额外的稳定性, 这与能量学分析结果一致.最后, 碱基的饱和体系中, 复合物的稳定性依然主要源于碱基-阴离子相互作用, 此时, lpBr-→ σN3—H*稳定能最高, 其次为lpBr-→ σN1—H*, 相较于T-[Cnmim]Br-1体系, 碱基与阳离子之间的∑Eπ→π+2数值增大, 依然高于∑Eπ←π+2, 离子对之间的轨道作用同样增加了体系的稳定性, 因此, T-[Cnmim]Br-1和T-2[Cnmim]Br-1体系中各片段两两间相互作用对复合物稳定化能的贡献大小遵循以下顺序: T-Br->[Cnmim]Br>T-[Cnmim]+.
| 表 5 M06-2X/6-311++G(2d, p)/PCM/water理论水平下基于复合物波函数信息的二阶稳定化能预测值 Table 5 The donor-acceptor interaction energies obtained from the NBO analysis on the wave functions for each complexe calculated at the M06-2X/6-311++G(2d, p)/PCM/water level |
1) 胸腺嘧啶与溴阴离子之间的相互作用强于该碱基与咪唑阳离子, 而阳离子烷基侧链长度对体系(T-[Cnmim]+、T-Br-、T-[Cnmim]Br、T-2[Cnmim]Br)稳定性的影响差异不大.
2) 胸腺嘧啶与阴离子之间以N—H…Br氢键相互作用为主, 与阳离子之间主要是ππ+堆积作用, 辅以C—H…O7或C—H…π弱氢键. N—H…Br氢键相互作用以静电吸引为主, 电子由溴离子转移至胸腺嘧啶, NBO轨道相互作用贡献最大的是lpBr→σN—H*. π…π+堆积作用以色散吸引为主, 电子主要由碱基转移至阳离子, NBO稳定化能的贡献主要源于π→π+*轨道相互作用.
3) 胸腺嘧啶与阴离子间形成两种作用模式, 共面型(front)和面上型(top);与阳离子间则是ππ+堆积(stacking)模式比垂直型(perpendicular)和共面型(coplanar)在能量学更有利.
3种其他碱基与溴化咪唑类离子液体的横纵比较计算分析结果也将陆续进行报道.
Araujo J M, Ferreira R, Marrucho I M, et al. 2011. Solvation of nucleobases in 1, 3-dialkylimidazolium acetate ionic liquids:NMR spectroscopy insights into the dissolution mechanism[J]. The Journal of Physical Chemistry B, 115(36): 10739-10749. |
Azizi A, Ebrahimi A. 2019. The effects of anion approaching directions to the π-π+ interaction[J]. Journal of Molecular Liquids, 276(2): 170-178. |
Bader R F W, Essén H J. 1984. The characterization of atomic interactions[J]. The Journal of Chemical Physics, 80(5): 1943-1960. |
Boys S F, Bernardi F. 2006. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors[J]. Molecular Physics:An International Journal at the Interface Between Chemistry and Physics, 19(4): 553-566. |
Bubalo M C, Radosevic K, Redovnikovic I R, et al. 2014. A brief overview of the potential environmental hazards of ionic liquids[J]. Ecotoxicology and Environmental Safety, 99(1): 1-12. |
Bubalo M C, Radosevic K, Redovnikovic I R, et al. 2017. Toxicity mechanisms of ionic liquids[J]. Arhiv Za Higijenu Rada I Toksikologiju, 68(3): 171-179. |
Bystrzanowska M, Pena-Pereira F. 2019. Marcinkowski, L, Tobiszewski, M. How green are ionic liquids?-a multicriteria decision analysis approach[J]. Ecotoxicology and Environmental Safety, 174(3): 455-458. |
曹建平, 牟永晓, 陈媛媛, 等. 2019. 离子液体在药物研究中的应用[J]. 药学学报, 54(2): 245-257. |
Cao L D, Zhu P, Zhao Y S, et al. 2018. Using machine learning and quantum chemistry descriptors to predict the toxicity of ionic liquids[J]. Journal of Hazardous Materials, 352: 17-26. |
Cauët E, Rooman M, Wintjens R, et al. 2005. Histidine-aromatic interactions in proteins and protein-ligand complexes:quantum chemical study of X-ray and model structures[J]. Journal of Chemical Theory and Computation, 1(3): 472-483. |
Chałasiński G. 2000. State of the art and challenges of the ab initio theory of intermolecular interactions[J]. Chemical Reviews, 100(11): 4227-4252. |
Chandran A, Ghoshdastidar D, Senapati S J. 2012. Groove binding mechanism of ionic liquids:A key factor in long-term stability of DNA in hydrated ionic liquids?[J]. Journal of the American Chemical Society, 134(50): 20330-20339. |
Corvo M C, Sardinha J, Menezes S C, et al. 2013. Solvation of carbon dioxide in[C4mim] [BF4] and [C4mim] [PF6] ionic liquids revealed by high-pressure NMR spectroscopy[J]. Angewandte Chemie International Edition, 52(49): 13024-13027. |
Crick F H C, Watson J D. 1954. The complementary structure of deoxyribonucleic acid[J]. Proceedings of the Royal Society A:Mathematical, Physical & Energeering Sciences, 223(8): 80-96. |
Dean P M, Pringle J M, MacFarlane D R. 2010. Structural analysis of low melting organic salts:perspectives on ionic liquids[J]. Physical Chemistry Chemical Physics, 12(32): 9144-9153. |
Ding Y H, Zhang L, Xie J, et al. 2010. Binding characteristics and molecular mechanism of interaction between ionic liquid and DNA[J]. Journal of Physical Chemistry B, 114(5): 2033-2043. |
董莹, 张淑娴, 刘惠君. 2015. 两种咪唑氯盐类离子液体对水稻幼苗根部的毒性效应[J]. 环境科学学报, 35(10): 3384-3389. |
Fan Y C, Dong X, Li X, et al. 2016. Spectroscopic studies on the inhibitory effects of ionic liquids on lipase activity[J]. Spectrochimica Acta Part A:Molecular and Biomolecular Spectroscopy, 159(4): 128-133. |
Fiethen A, Jansen G, Hesselmann A, et al. 2008. Stacking energies for average B-DNA structures from the combined density functional theory and symmetry-adapted perturbation theory approach[J]. Journal of the American Chemical Society, 130(6): 1802-1803. |
Frisch M J, Trucks G W, Schlegel H B, et al. 2016. Gaussian 09 Revision D.01 Gaussian Inc, Wallingford CT
|
Glendening E D, Landis C R, Weinhold F. 2013. NBO 6.0:Natural bond orbital analysis program[J]. Journal of Computational Chemistry, 34(16): 1429-1437. |
Gu J, Wang J, Leszczynski J. 2011. Stacking and H-bonding patterns of dGpdC and dGpdCpdG:Performance of the M05-2X and M06-2X Minnesota density functionals for the single strand DNA[J]. Chemical Physics Letters, 512(1/3): 108-112. |
Hong B H, Lee J Y, Lee C W, et al. 2001. Self-assembled arrays of organic nanotubes with infinitely long one-dimensional H-bond chains[J]. Journal of the American Chemical Society, 123(43): 10748-10749. |
Hunt P A, Ashworth C R, Matthews R P. 2015. Hydrogen bonding in ionic liquids[J]. Chemical Society Reviews, 44(5): 1257-1288. |
Jumbri K, Micaelo N M, Abdul Rahman M B. 2016. Solvation free energies of nucleic acid bases in ionic liquids[J]. Molecular Simulation, 43(1): 19-27. |
Jumbri K, Rahman M B A, Abdulmalek E, et al. 2014. An insight into structure and stability of DNA in ionic liquids from molecular dynamics simulation and experimental studies[J]. Physical Chemistry Chemical Physics, 16(27): 14036-14046. |
Karimi P. 2019. Investigation of intramolecular hydrogen bonding in conjunction with cation-π interactions in complexes involving 4-substituted-8-hydroxyquinolines[J]. Journal of Molecular Structure, 1182(4): 266-270. |
Kasende O, E, Nziko Vde P, Scheiner S. 2016. Interactions of nucleic acid bases with temozolomide. stacked, perpendicular, and coplanar heterodimers[J]. The Journal of Physical Chemistry B, 120(35): 9347-9361. |
Kataev E A, Shumilova T A, Fiedler B, et al. 2016. Understanding stacking interactions between an aromatic ring and nucleobases in aqueous solution:experimental and theoretical study[J]. The Journal of Organic Chemistry, 81(15): 6505-6514. |
Koch U, Popelie P L A. 1995. Characterization of C-H-O hydrogen bonds on the basis of the charge density[J]. The Journal of Physical Chemistry, 99(24): 9747-9754. |
Lefebvre C, Rubez G, Khartabil H, et al. 2017. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density[J]. Physical Chemistry Chemical Physics, 19(27): 17928-17936. |
Leontis N B, Stombaugh J, Westhof E. 2002. The non-Watson-Crick base pairs and their associated isostericity matrices[J]. Nucleic Acids Research, 30(16): 3497-3531. |
刘宝友, 张佩文. 2018. 离子液体的进展——绿色制备及在环境修复中的应用研究[J]. 有机化学, 38(12): 3176-3188. |
Nakano M, Tateishi-Karimata H, Tanaka S, et al. 2014. Choline ion interactions with DNA atoms explain unique stabilization of A-T base pairs in DNA duplexes:a microscopic view[J]. The Journal of Physical Chemistry B, 118(2): 379-389. |
Parthasarathi R, Subramanian V, Sathyamurthy N. 2006. Hydrogen bonding without borders:an atoms-in-molecules perspective[J]. The Journal of Physical Chemistry A, 110(10): 3349-3351. |
Petti M A, Shepodd T J, Barrans R E, et al. 1988. "Hydrophobic" binding of water-soluble guests by high-symmetry, chiral hosts. An electron-rich receptor site with a general affinity for quaternary ammonium compounds and electron-deficient π systems[J]. Journal of the American Chemical Society, 110(20): 6825-6840. |
Pham T P T, Cho C W, Yun Y S. 2010. Environmental fate and toxicity of ionic liquids:A review[J]. Water Research, 44(2): 352-372. |
Popelier P L A. 1998. Characterization of a dihydrogen bond on the basis of the electron density[J]. The Journal of Physical Chemistry A, 102(10): 1873-1878. |
Popelier P L A. 2000. On the full topology of the Laplacian of the electron density[J]. Coordination Chemistry Reviews, 197(6): 169-189. |
Pullman A, Perahia D. 1978. Hydration scheme of uracil and cytosine[J]. Theoretica Chimistry Accounts, 48(1): 29-36. |
Qu R J, Liu H X, Feng M B, et al. 2012. Investigation on intramolecular hydrogen bond and some thermodynamic properties of polyhydroxylated anthraquinones[J]. Journal of Chemical & Engineering Data, 57(9): 2442-2455. |
Reed A E, Weinstock R B, Weinhold F J. 1985. Natural population analysis[J]. The Journal of Chemical Physics, 83(2): 735-746. |
Ricca A, Bauschlicher C W. 1995. Successive H2O binding energies for Fe(H2O)n+[J]. The Journal of Physical Chemistry, 99(22): 9003-9007. |
Tateishi-Karimata H, Sugimoto N, Biophys R. 2018. Biological and nanotechnological applications using interactions between ionic liquids and nucleic acids[J]. Biophysical Reviews, 10(3): 931-940. |
Vijay D, Sastry G N. 2010. The cooperativity of cation-π and π-π interactions[J]. Chemical Physics Letters, 485(12): 235-242. |
Vijay D, Zipse H, Sastry G N. 2008. On the cooperativity of cation-π and hydrogen bonding interactions[J]. Journal of Physical Chemistry B, 112(30): 8863-8867. |
Vraneš M, Tot A, Jovanović-Šanta S, et al. 2016. Toxicity reduction of imidazolium-based ionic liquids by the oxygenation of the alkyl substituent[J]. RSC Advances, 6(98): 96289-96295. |
Walker M, Harvey A J, Sen A, et al. 2013. Performance of M06, M06-2X, and M06-HF density functionals for conformationally flexible anionic clusters:M06 functionals perform better than B3LYP for a model system with dispersion and ionic hydrogen-bonding interactions[J]. The Journal of Physical Chemistry A, 117(47): 12590-12600. |
Wang C, Wei Z B, Feng M B, et al. 2014. Comparative antioxidant status in freshwater fish Carassius auratus exposed to eight imidazolium bromide ionic liquids:A combined experimental and theoretical study[J]. Ecotoxicology and Environmental Safety, 102: 187-195. |
Wang C, Wei Z B, Wang L S, et al. 2015. Assessment of bromide-based ionic liquid toxicity toward aquatic organisms and QSAR analysis[J]. Ecotoxicology and Environmental Safety, 115: 112-118. |
Wang H Y, Wang J J, Zhang S B. 2011. Binding Gibbs energy of ionic liquids to calf thymus DNA:A fluorescence spectroscopy study[J]. Physical Chemistry Chemical Physics, 13(9): 3906-3910. |
王引航, 李伟, 罗沙, 等. 2018. 离子液体固载型功能材料的应用研究进展[J]. 化学学报, 76(2): 85-94. |
Wu S G, Zeng L B, Wang C Y, et al. 2018. Assessment of the cytotoxicity of ionic liquids on Spodoptera frugiperda 9 (Sf-9) cell lines via in vitro assays[J]. Journal of Hazardous Materials, 348(4): 1-9. |
徐佳禛, 牟永晓, 曹建平, 等. 2018. 溴化咪唑类离子液体对胞嘧啶结构和性质影响的计算研究[J]. 环境科学学报, 38(10): 4113-4123. |
Xu J Z, Yi L G, Mou Y X, et al. 2018. Effect of a molecule of imidazolium bromide ionic liquid on the structure and properties of cytosine by density functional theory[J]. Chemical Physics Letters, 708(9): 109-116. |