 2020, Vol. 40
2020, Vol. 40



2. 华中师范大学化学学院, 农药与化学生物学教育部重点实验室, 武汉 430079
2. Laboratory of Pesticide & Chemical Biology of Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079
过硫酸盐是一类常见的氧化剂, 可用作漂白剂、氧化剂、乳液聚合促进剂, 并在照相工业、纺织工业、石油开采等领域有着广泛的应用(杨世迎等, 2008).而过硫酸盐的活化作为高级氧化技术(Advanced Oxidation Process, AOPs)的一种, 在环境污染治理领域已得到了非常广泛的发展.
过硫酸盐的活化机制主要包含热、光(Hori et al., 2005)、微波(Qi et al., 2015; Hamid et al., 2018)、碱(Furman et al., 2010)、金属离子(Lee et al., 2010; Jo et al., 2014)、活性炭活化(Lee et al., 2013; Wang et al., 2018)等方式.而超声也被视作热活化方法的一种, 因为在超声过程中空化气泡的破裂会产生局部高温(Lin et al., 2015).经活化后的过硫酸盐可产生强氧化性的SO4·-(E0=2.6 V vs NHE), 进而可以降解环境污染物(Yu et al., 2016; Zhang et al., 2018).
以全氟辛酸(PFOA)为代表的全氟化合物是一类新兴的环境持久性有机污染物, 具有环境持久性、生物累积性和高毒性等, 因而引起了全球范围内的广泛研究(Poothong et al., 2012).已有众多研究显示, PFOA与出生缺陷(Manzano et al., 2017)、肝细胞凋亡(Li et al., 2017)、肾脏毒性(Kudo et al., 2003)、代谢综合征(Yang et al., 2018)等疾病有关, 对人类的生命健康存在着严重的威胁.迄今为止还尚未发现PFOA等自然降解的途径, 而许多传统的高级氧化技术如紫外光直接降解、生物降解、·OH和H2O2/Fe2+等均无法实现对其有效的降解.因此, 寻找高效的全氟辛酸降解方法成为环境领域学者们关注的热点.例如, Hori等(2004)发现用钨杂多酸光催化剂(H3PW12O40·6H2O)可以有效光化学降解水中的PFOA, 反应24 h后的脱氟率为88%.Shang等(2018)报道了一种基于混合氧化物的新型钙钛矿型光催化剂(Pb-BiFeO3)对PFOA的光催化降解, 反应8 h后降解率达69.6%, 脱氟率可达37.6%.另外, 利用Fe3+与PFCAs在紫外光辐射下发生配体-金属电子转移(LCMT)反应来实现对PFOA的降解研究也有所报道(Tang et al., 2012; Yuan et al., 2020).
由于电化学降解法具有操作简单、条件可控和降解高效的特点, 近年来在有机废水处理方面受到了高度的关注(Zhuo et al., 2011).目前, 应用电化学技术降解全氟化合物的研究多集中在研制具有较高析氧电位和疏水性强的新型电化学复合电极方面, 从而实现对PFOA的电化学直接氧化降解(Lin et al., 2010; Lin et al., 2012).此时电极的制备步骤通常都很繁琐和复杂.有文献报道在UV/S2O82-光化学体系中, 利用硫酸根自由基(SO4·-)可实现对PFOA的氧化降解, 然而该方法能耗较大.为此, 本文结合高级氧化与电化学技术, 利用经济易得的多孔碳毡电极去电活化过硫酸盐, 实现由SO4·-介导的电化学降解PFOA的过程.同时, 探讨恒定电位值、过硫酸钠初始浓度及溶液初始pH值等对电活化过硫酸盐降解PFOA的影响.另外, 通过模拟自然场景, 对水溶液中可能的共存离子对PFOA降解效果的影响进行考察.最后综合获得的TOC去除率和降解中间产物等实验结果, 提出可能的反应机理.以期为全氟辛酸在低能耗下的矿化提供新的可能, 并为中间产物的探索提供新的方向.
2 材料与方法(Materials and methods) 2.1 主要仪器和试剂试剂:多孔碳毡CF(纯度>98%, 电阻率0.3 Ω·cm, 厚度3 mm, 密度0.09 g·cm-2, 浩特新材料)、过硫酸钠(分析纯, 国药)、全氟辛酸(分析纯, 阿拉丁试剂).
仪器:电化学工作站(CS350, 武汉科斯特有限公司)、超高效液相色谱-三重四级杆串联质谱联用仪(安捷伦科技有限公司)、离子色谱(Aquion, Dionex, USA, 赛默飞世尔科技有限公司)、总有机碳分析仪(CPH, 日本岛津有限公司).
2.2 全氟辛酸的降解实验移取10 mL 100 mg·L-1 PFOA标准溶液于200 mL烧杯中, 向其中加入190 mL超纯水, 将其稀释成200 mL 5 mg·L-1的PFOA溶液, 作为全氟辛酸模拟污水溶液.
向所得溶液中加入2.3810 g过硫酸钠(Na2S2O8), 手动搅拌使之充分溶解, 使溶液中过硫酸钠浓度为50 mmol·L-1.将已清洗干净并充分干燥的多孔碳毡电极(CF电极)用镊子夹好, 完全浸入到所得溶液中, 用封口膜将烧杯口封好并固定, 用空气压缩机鼓气, 吸附过夜, 使多孔碳毡电极在该浓度的PFOA溶液中达到吸附-脱附平衡状态.
接下来移取2.5 mL 100 mg·L-1的PFOA标准溶液于50 mL的聚乙烯烧杯中, 加入47.5 mL超纯水, 将其稀释成50 mL 5 mg·L-1的PFOA溶液, 作为全氟辛酸模拟污水溶液.之后加入0.5953 g过硫酸钠(Na2S2O8), 手动搅拌使之充分溶解, 使溶液中初始过硫酸钠的浓度为50 mmol·L-1, 与吸附时保持一致.
将所得的50 mL的5 mg·L-1 PFOA与50 mmol·L-1 Na2S2O8的混合溶液作为电解质溶液, 用已达吸附-脱附平衡的CF电极(1 cm×2 cm)作阴极(同时作为工作电极), 铂片(1 cm2)作阳极(同时作为参比电极和对电极), 搭建电化学降解全氟辛酸的装置.通过电化学工作站(Correst-CS350)给工作电极(CF电极)施加一定电位, 鼓气, 采用恒电位法降解全氟辛酸.按一定时间间隔取样, 每次取样1 mL, 用直径13 mm、孔径0.22 μm的有机针筒过滤器过滤, 考察模拟全氟辛酸污水溶液中PFOA的电化学去除情况.
加入适量乙醇来同时捕获体系中的硫酸根自由基和羟基自由基, 加入叔丁醇单独捕获体系中的羟基自由基, 用以研究不同自由基对于全氟辛酸降解过程的影响.
2.3 分析方法样品溶液需先经过直径13 mm、孔径0.22 μm的有机针筒过滤器过滤, 其中, PFOA的浓度全部通过超高效液相色谱-三重串联四级杆质谱联用仪(Agilent 1290 UPLC-6460 QQQ MSD)进行测定.所用色谱柱为:Agilent ZORBAX Eclipse Plus C18(3.0 mm×100 mm, 3.5 μm).液相条件为:进样体积为10 μL, 流动相为70%的乙酸铵(CH3COONH4, 10 mmol·L-1)和30%的乙腈, 流速为0.3 mL·min-1, 柱温箱温度为40 ℃, 高真空在2×105 Torr;质谱条件为:ESI源, 负离子模式, 毛细管电压为4000 V, 喷嘴电压为500 V, 鞘气温度为350 ℃, 鞘气流速为10 L·min-1, 载气(氮气)压力为45 psi, 流速为6 L·min-1, 碰撞气(氮气)温度为325 ℃, 多反应监测(MRM)模式.测定反应中间产物过程中, 全扫时采用Fullscan模式.全氟羧酸母离子(m/z)、子离子(m/z)、保留时间(min)参数分别为:PFOA(413>369.5, 5.901)、PFHpA(363>319.5, 5.000)、PFHxA(313>269.3, 3.952)、PFPeA(263>219.2, 2.700).
反应过程自由基通过电子顺磁共振波谱仪监测(Bruker, EMXmicro-6/1), F的去向分别采用离子色谱仪(Aquion, Dionex, USA)测定, 反应溶液中总有机碳(TOC)含量的变化通过总有机碳分析仪(CPH)进行检测.
反应的中间产物分别通过超高效液相色谱-三重串联四级杆质谱联用仪(UPLC-MS/MS)和气相色谱-质谱联用仪(GC-MS)进行测定.另外, 在对气相产物的收集中, 分别采用可复溶回反应溶液的气相产物法(Hori et al., 2006)、顶空吹扫捕集法和顶空直接进样法收集, 结果再同GC-MS数据库进行匹配.反应过程中总有机碳(TOC)含量通过总有机碳分析仪(CPH)测定.
3 结果与讨论(Results and discussion) 3.1 不同因素对PFOA降解效果的影响为能有效地电化学活化过硫酸盐, 采用多孔碳毡电极作为工作电极, 铂片电极作为对电极, 饱和甘汞电极作为参比电极, 以过硫酸钠溶液(0.05 mol·L-1)为支持电解质, 测定了该体系的循环伏安曲线(图 1a).可以看到在电压约为-1.15 V时, S2O82-可以得电子还原生成SO4·-(式(1)).另外, 从电子自旋共振谱图(图 1b)中可知, 反应体系中存在着SO4·-和·OH.

|
(1) |
 |
| 图 1 0.05 mol·L-1过硫酸钠溶液(Na2S2O8)的循环伏安曲线(a)及经电解30 min后(E = -1.15 V)的电子自旋共振谱图(EPR)(b) Fig. 1 Cyclic voltammogram of 0.05 mol·L-1 Na2S2O8 solution(a) and EPR spectra of 0.05 mol·L-1 Na2S2O8 solution under the electrolysis of E = -1.15 V for 30 min(b) |
实验选取恒电位的考察值分别为-1.2、-1.8、-2.0、-3.0和-5.0 V, 此时PFOA的降解情况如图 2a所示.可以看到随着设置的恒定电位值逐渐变负, PFOA的降解效率逐渐提高.当恒定电位值为-1.2 V时, 反应4 h后PFOA约降解了40%, 而继续减小电位至-4.0 V时, 反应4 h后PFOA的降解效率约达到了90%, 此时可能是大量的PFOA在阴极上直接被还原.另外, 考虑到过高的电位会引起水的电解, 导致降解体系不稳定.因此, 后续实验均采用较为温和的-1.8 V电位进行实验.发现当E=-1.8 V, 延长反应时间, 在电解约8 h时, PFOA的去除率已接近100%(图 2b).
 |
| 图 2 恒定电位值(a)对PFOA降解效果的影响、-1.8 V电位下PFOA的降解效果(b)及过硫酸盐浓度(c)和反应溶液初始pH(d)对PFOA降解效果的影响 Fig. 2 Effect of constant potential on degradation of PFOA(a), time dependence of PFOA decomposition under-1.8 V(b), effect of the concentration(c) and initial pH value(d) of persulfate on degradation of PFOA |
如图 2c所示, 在一定范围内, PFOA的降解效率随着过硫酸钠浓度的增大而升高, 超过200 mmol·L-1后, 随着过硫酸盐浓度的升高, 降解效果反而下降.这可归结为S2O82-与SO4·-自由基的猝灭作用(式(2))(Bekirs et al., 2017).因此, 在后续实验中采用200 mmol·L-1过硫酸钠溶液作为电解液进行电化学降解实验.
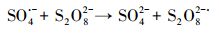
|
(2) |
由于反应过程中H+的浓度可能会引起活性物种的变化, 进而影响到降解效果(式(3)和(4))(Lee et al., 2012; Yin et al., 2016), 因此, 对反应溶液的初始pH进行了优化.
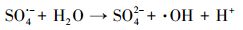
|
(3) |
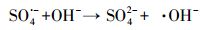
|
(4) |
由反应式(3)可以看出, 酸性条件有利于体系中SO4·-的稳定存在.同时, 由于在强酸性条件下对于过硫酸根转化成硫酸根自由基的酸催化作用也可促进SO4·-的生成.另外, 碱性条件下大量存在的OH-会消耗SO4·-(式(4)).为此, 在此过程中只考虑了在接近其初始pH(pH=3.29)的酸性范围(pH=2.0~5.0)下进行pH的优化, 未考虑碱性条件对反应体系的影响.
由反应结果图(图 2d)可以看出, 当溶液pH为2时, PFOA的降解受到了抑制, 这是由于在此pH下阴极的析氢反应剧烈, 不利于S2O82-在阴极上的活化.因此, 后续采用初始pH=3.29的溶液进行实验.
3.1.4 共存离子对降解效果的影响为了模拟自然界中多种离子共存的环境, 以及探讨其他阴离子与过硫酸的混合体系对PFOA降解效果的影响, 设计了5组共存离子(分别为NO3-、NO3-+异丙醇、HCO3-、Cl-和ClO4-)实验.
如图 3a所示, 加入硝酸盐的两组之中, 仅加硝酸盐的一组其降解效果与单独过硫酸钠体系几乎相似.在体系中进一步引入异丙醇(·OH捕获剂), 发现其对PFOA的降解效果有了显著的提升, 在电解4 h后降解效率已达到91%.这可能归结为当捕获·OH之后, 体系中产生了大量的·NO2自由基与强氧化性的SO4·-协同作用, 从而大幅提升了PFOA的去除速率.这与本课题组之前报道的UV/NO3-降解PFOA体系的结论相一致(Li et al., 2017).在引入NaHCO3时, PFOA的降解效果受到极大的抑制, 这是由于HCO3-会猝灭SO4·-; 另外, 引入HCO3-也会导致溶液pH的上升, 不利于SO4·-的稳定(式(5)、(4)).而加入NaCl的一组呈现出一定的抑制效果, 这可能是因为发生了如下反应(式(6)), 氯离子的引入消耗了在PFOA降解过程中起主要作用的活性物种SO4·-, 使得其降解效果受到一定的抑制.
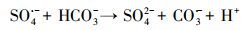
|
(5) |
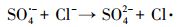
|
(6) |
 |
| 图 3 共存离子(100 mmol·L-1)对PFOA降解效果的影响(a)及反应体系中总有机碳浓度的变化(b) Fig. 3 Effect of coexisting ions(100 mmol·L-1) on degradation of PFOA(a) and time dependence of the removal of TOC in solution(b) |
加入NaClO4的一组表现出一定的促进作用, 电解4 h后PFOA降解率达到76.8%.这可能是因为NaClO4在电解过程中会在电极上发生氧化还原反应, 生成具有强氧化性的多种活性氯物种(如Cl2·-、HOCl/ClO-和ClO3-等), 从而促进PFOA的降解.
3.2 电化学活化过硫酸盐降解PFOA的产物研究 3.2.1 总有机碳(TOC)及离子色谱的测定为了探究电活化过硫酸钠降解PFOA的机理, 首先测定了反应溶液中总有机碳(TOC)含量随时间的变化并计算出总有机碳的去除率.结果表明, 在反应24 h后, TOC的去除率达到62.5%(图 3b).
而对反应24 h之后的样品进行离子色谱测定, 测得溶液中F-的含量仅为0.7170 mg·L-1, 计算得到脱氟率约为20.84%, 表明在反应24 h之后未能实现PFOA的完全矿化, 而且还有一部分的产物存在于气相中.
3.2.2 降解反应中间产物的分析首先使用UPLC-MS/MS的多反应监控(MRM)模式对中间产物进行了测定.从色谱图(图 4)可以看出, 降解反应中生成了短链的全氟羧酸(全氟戊酸PFPeA、全氟己酸PFHxA和全氟庚酸PFHpA), 证明降解过程中存在传统的Kolbe脱羧(—CF2)过程, 中间产物相对应的保留时间如表 1所示.
 |
| 图 4 电解反应4 h后反应的中间产物示意图 Fig. 4 The chromatogram of intermediate products after 4 h |
| 表 1 中间产物及其对应保留时间 Table 1 Intermediate products and retention time |
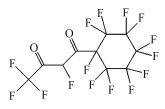
参考Huang等(2016)的研究, 从UPLC-MS/MS-Full scan模式测定质谱图中发现, 除PFHpA, PFHxA和PFPeA的碎片离子峰之外, 还找到了一种α-C上一氢取代的全氟庚酸CF3(CF2)4CFHCOOH、一氢取代的全氟丁烷CF3(CF2)2CF2H、两氢取代的全氟己烷CF3(CF2)4CFH2、一氢取代的全氟己烯CF2=CF(CF2)3CF2H, 对应产物及其保留时间如表 2所示.
| 表 2 全扫模式检测到的中间产物 Table 2 Intermediate products detected in full scan mode |
由于前两种方法检测出的中间产物的量较少和较低的脱氟率, 故推测在反应过程中, 有一部分中间产物可能转化为气相形式存在.因此, 进一步采用GC-MS进行气相产物的检测.
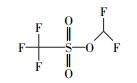
通过与GC-MS数据库的对比, 从顶空直接进样法中匹配到了极短链的全氟羧酸(全氟乙酸CF3COOH)、全氟甲酸乙酯FCOOC2H5、二氢取代的全氟乙酸CH2FCOOH及一氟取代的丙烷和异丁烷.结果表明, 在降解PFOA的过程中除了典型的Kolbe脱羧过程中得到的短链全氟羧酸中间产物外, 还有加氢还原及气相产物的存在(表 3).
| 表 3 顶空直接进样法气相产物结构示意图 Table 3 Schematic diagram of the structure of gas-phase products by headspace direct sampling |
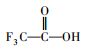
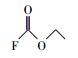

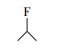
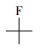
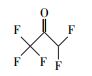
从可复溶回反应溶液的气相产物法中匹配到了全氟烷烃(全氟乙烷(C2F6)、全氟己烷(C6F14)等)、一氢取代的全氟烷烃(一氢取代的全氟甲烷(CF3H)、一氢取代的全氟乙烷(C2F5H)和一氢取代的全氟己烷(C6F13H))、全氟醛类(全氟丁醛(C3F7CHO)和全氟辛醛(C7F15CHO))、一氢取代的全氟醛类(全氟戊醛、全氟己醛、全氟庚醛)、全氟醇类(全氟庚醇C6F13CH2OH)、一氢取代的全氟酮、醚等, 以及一种未知的C、H、F、O复合物(表 4).
| 表 4 可溶性物质复溶法气相产物结构示意 Table 4 Schematic diagram of gas-phase products structure by reconstitution of soluble substance |
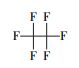
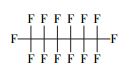
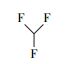
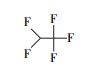
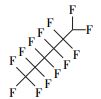
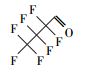
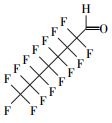
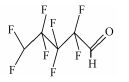
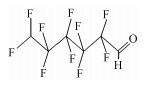
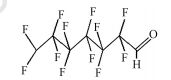
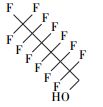
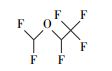
从吹扫捕集法中匹配到了多种不饱和的含氟烃类, 如1, 1-二氟乙烯、1, 2-二氟乙烯、3, 3, 4, 4-四氟-1, 5-己二烯和1, 4-二氟-1, 3-丁二炔等.此外, 还有全氟甲醛、1, 1-二氟乙烷、1, 2-二氟乙烷、二氢取代的全氟乙酸CH2FCOOH等含F物质生成(表 5).
| 表 5 顶空吹扫捕集法气相产物结构示意图 Table 5 Schematic diagram of gas-phase products structure by purge and trap |
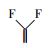
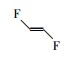
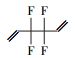
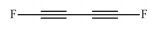
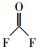
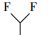
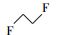
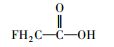
为推测电化学活化过硫酸盐降解PFOA可能的反应机理, 首先进行了不同电解质溶液的对比实验和自由基的捕获实验.
为了验证过硫酸钠中S2O82-的作用, 分别考察了相同浓度的Na2SO4和Na2S2O8在相同电位下对于PFOA的降解情况.结果表明, 在以Na2SO4为电解质溶液时, 反应8 h后PFOA的降解效率仅为20%, 而在Na2S2O8的电解体系中PFOA的降解效率可达100%, 说明S2O82-对降解PFOA的确起到了作用(图 5a).
 |
| 图 5 不同电解质对比实验(a)及自由基捕获实验(b) Fig. 5 The degradation of PFOA in different electrolytes(a) and the degradation effect of PFOA after adding radical scavenger(b) |
另外, 有文献报道, 叔丁醇可用作单独的·OH捕获剂, 而乙醇可同时捕获·OH和SO4·-(Qi et al., 2015).因此, 本文分别考察了引入叔丁醇和乙醇情况下对PFOA降解的影响.如图 5b所示, 两种捕获剂均对降解效果有抑制作用, 加入叔丁醇后PFOA的降解率为43%, 抑制率约为9%;加入乙醇后PFOA的降解率为23.5%, 抑制率约为50%, 表明SO4·-和·OH均能促进PFOA的降解且SO4·-发挥主要作用(图 5b).
根据之前的中间产物分析和活性物种的作用, 推测电化学活化过硫酸钠降解PFOA的过程可分为两部分:①一部分是碳链减少的Kolbe脱羧过程:S2O82-得电子被还原成SO4·-, 而具有强氧化性的SO4·-可参与到反应中, 促进PFOA降解反应第一步的发生, 即PFOA分子失去电子, 形成全氟辛酸自由基(C7F15COO·), 再经过Kolbe脱羧, 形成全氟庚基自由基(C7F15·); 随后其与·OH反应生成C7F15OH, 脱去HF生成C7F13OF, 再经水解脱去HF生成C6F13COO-.
② 另一部分是PFOA在阴极上直接得电子的羧酸逐步被加氢还原的过程:由全氟羧酸加氢形成一氢取代的全氟羧酸, 再还原成醛、醇, 消去生成烯烃, 最后氢加成生成一氢或二氢取代的全氟烷烃, 或与氟加成形成全氟烷烃.相应的产物同一些之前的研究相吻合(Vecetis et al., 2008; Qu et al., 2010).具体机理如图 6所示.
 |
| 图 6 电化学活化过硫酸盐降解PFOA的机理(机理I:碳链减少的Kolbe脱羧过程;机理II:加氢还原过程) Fig. 6 The mechanism of electrochemical degradation of perfluorooctanoic acid by electrochemical activation of persulfate(Mechanism I: the process of reduction of carbon chains length. Mechanism II: the process of hydrogenation reduction) |
本文用多孔碳毡电极作为工作电极, 用恒电位法活化过硫酸降解PFOA, 探究了反应过程中恒电位大小、过硫酸盐初始浓度、反应溶液pH值、共存离子对降解效果的影响, 并监测了反应过程中的PFOA降解率、有机相、气相产物、矿化率等.结果表明, 在-1.8 V的恒电位下电解约8 h后PFOA去除率已接近100%.而延长反应时间至24 h, 体系中的总有机碳(TOC)含量下降了约62.5%.同时, 利用GC-MS对气相产物进行了鉴定, 推断其可能的降解机理为SO4·-介导的Kolbe脱羧过程和羧酸逐步被加氢还原的过程.此方法为全氟辛酸在低能耗下的降解提供了新的可能, 也为中间产物的探索提供了新的方向.
Bekris L, Frontistis Z, Trakakis G, et al. 2017. Graphene:a new activator of sodium persulfate for the advanced oxidation of parabens in water[J]. Water Research, 126(1): 111-121. |
Furman O S, Teel A L, Watts R J. 2010. Mechanism of base activation of persulfate[J]. Environmental Science & Technology, 44(16): 6423-6428. DOI:10.1021/es1013714 |
Hamid H, Li L. 2018. Fate of perfluorooctanoic acid (PFOA) in sewage sludge during microwave-assisted persulfate oxidation treatment[J]. Environmental Science and Pollution Research, 25: 10126-10134. DOI:10.1007/s11356-018-1576-3 |
Hori H, Hayakawa E, Einaga H, et al. 2004. Decomposition of environmentally persistent perfluorooctanoic acid in water by photochemical approaches[J]. Environmental Science & Technology, 38(22): 6118-6124. |
Hori H, Nagaoka Y, Yamamoto A, et al. 2006. Efficient decomposition of environmentally persistent perfluorooctanesulfonate and related fluorochemicals using zerovalent iron in subcritical water[J]. Environmental Science & Technology, 40(3): 1049-1054. |
Hori H, Yamamoto A, Hayakawa E, et al. 2005. Efficient decomposition of environmentally persistent perfluorocarboxylic acids by use of persulfate as a photochemical oxidant[J]. Environmental Science & Technology, 39(7): 2383-2388. |
Huang D, Yin L, Niu J. 2016. Photoinduced hydrodefluorination mechanisms of perfluorooctanoic acid by the SiC/graphene catalyst[J]. Environmental Science & Technology, 50(11): 5857-5863. |
Jo Y, Do S, Kong S. 2014. Persulfate activation by iron oxide-immobilized MnO2 composite:identification of iron oxide and the optimum pH for degradations[J]. Chemosphere, 95: 550-555. DOI:10.1016/j.chemosphere.2013.10.010 |
Kudo N, Kawashima Y J. 2003. Toxicity and toxic kinetics of perfluorooctanoic acid in humans and animals[J]. The Journal of Toxicological Sciences, 28(2): 49-57. DOI:10.2131/jts.28.49 |
Lee Y, Lo S, Chiueh P, et al. 2010. Microwave-hydrothermal decomposition of perfluorooctanoic acid in water by iron-activated persulfate oxidation[J]. Water Research, 44(3): 886-892. DOI:10.1016/j.watres.2009.09.055 |
Lee Y, Lo S, Kuo J, et al. 2013. Promoted degradation of perfluorooctanic acid by persulfate when adding activated carbon[J]. Journal of Hazardous Materials, 261: 463-469. |
Lee Y, Lo S, Kuo J, et al. 2012. Persulfate oxidation of perfluorooctanoic acid under the temperatures of 20~40℃[J]. Chemical Engineering Journal, 198-199: 27-32. DOI:10.1016/j.cej.2012.05.073 |
Li A, Zhang Z, Li P, et al. 2017. Nitrogen dioxide radicals mediated mineralization of perfluorooctanoic acid in aqueous nitrate solution with UV irradiation[J]. Chemosphere, 188: 367-374. DOI:10.1016/j.chemosphere.2017.08.170 |
Lin J, Lo S, Hu C, et al. 2015. Enhanced sonochemical degradation of perfluorooctanoic acid by sulfate ions[J]. Ultrasonics Sonochemistry, 2: 542-547. |
Li K, Sun J, Yang J, et al. 2017. Molecular mechanisms of perfluorooctanoate-induced hepatocyte apoptosis in mice using proteomic techniques[J]. Environmental Science & Technology, 51(19): 11380-11389. |
Lin H, Niu J, Ding S, et al. 2010. Electrochemical degradation of perfluorooctanoic acid (PFOA) by Ti/SnO2-Sb, Ti/SnO2-Sb/PbO2 and Ti/SnO2-Sb/MnO2 anodes[J]. Water Research, 46(7): 2281-2289. |
Lin H, Niu J, Xu J, et al. 2013. Highly efficient and mild electrochemical mineralization of long-chain perfluorocarboxylic acids (C9-C10) by Ti/SnO2-Sb-Ce, Ti/SnO2-Sb/Ce-PbO2, and Ti/BDD electrodes[J]. Environmental Science & Technology, 47(22): 13039-13046. |
Manzano-Salgado C, Casas M, Lopez-Espinosa M, et al. 2017. Prenatal exposure to perfluoroalkyl substances and birth outcomes in a Spanish birth cohort[J]. Environment international, 108: 278-284. DOI:10.1016/j.envint.2017.09.006 |
Poothong S Boontanon, S K Boontanon N. 2012. Determination of perfluorooctane sulfonate and perfluorooctanoic acid in food packaging using liquid chromatography coupled with tandem mass spectrometry[J]. Journal of Hazardous Materials, 205-206: 139-143. DOI:10.1016/j.jhazmat.2011.12.050 |
Qi C, Liu X, Lin C, et al. 2015. Degradation and dechlorination of pentachlorophenol by microwave activated persulfate[J]. Environmental Science & Pollution Research International, 22(6): 4670-4679. |
Qu Y, Zhang C, Li F, et al. 2010. Photo-reductive defluorination of perfluorooctanoic acid in water[J]. Water Research, 44(9): 2939-2947. DOI:10.1016/j.watres.2010.02.019 |
Shang E, Li Y, Niu J, et al. 2018. Photocatalytic degradation of perfluorooctanoic acid over Pb-BiFeO3/rGO catalyst:kinetics and mechanism[J]. Chemosphere, 211: 34-43. DOI:10.1016/j.chemosphere.2018.07.130 |
Tang H, Xiang Q, Lei M, et al. 2012. Efficient degradation of perfluorooctanoic acid by UV-Fenton process[J]. Chemical Engineering Journal, 184: 156-162. DOI:10.1016/j.cej.2012.01.020 |
Vecetis C, Park H, Cheng J, et al. 2008. Kinetics and mechanism of the sonolytic conversion of the aqueous perfluorinated surfactants, perfluorooctanoate(PFOA), and perfluorooctane sulfonate(PFOS) into inorganic products[J]. The Journal of Physical Chemistry A, 112(18): 4261-4270. DOI:10.1021/jp801081y |
Wang J, Wang S. 2018. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants[J]. Chemical Engineering Journal, 334: 1502-1517. DOI:10.1016/j.cej.2017.11.059 |
Yang Q, Guo X, Sun P, et al. 2018. Association of serum levels of perfluoroalkyl substances (PFASs) with the metabolic syndrome (MetS) in Chinese male adults:a cross-sectional study[J]. Science of the Total Environment, 621: 1542-1549. DOI:10.1016/j.scitotenv.2017.10.074 |
杨世迎, 陈友媛, 胥慧真, 等. 2008. 过硫酸盐活化高级氧化新技术[J]. 化学进展, 20(9): 1433-1438. |
Yin P, Hu Z, Song X, et al. 2016. Activated persulfate oxidation of perfluorooctanoic acid (PFOA) in groundwater under acidic conditions[J]. International Journal of Environmental Research and Public Health, 13(6): 602. DOI:10.3390/ijerph13060602 |
Yu Y, Zhou S, Bu L, et al. 2016. Degradation of diuron by electrochemically activated persulfate[J]. Water, Air, & Soil Pollution, 227(8): 279. |
Yuan Y, Feng Li, Zhang Z, et al. 2020. Rapid photochemical decomposition of perfluorooctanoic acid mediated by a comprehensive effect of nitrogen dioxide radicals and Fe3+/Fe2+ redox cycle[J]. Journal of Hazardous Materials, 388: 121730. DOI:10.1016/j.jhazmat.2019.121730 |
Zhang L, Ding W, Qiu W, et al. 2018. Modeling and optimization study on sulfamethoxazole degradation by electrochemically activated persulfate process[J]. Journal of Cleaner Production, 197: 297-305. DOI:10.1016/j.jclepro.2018.05.267 |
Zhuo Q, Deng S, Yang B, et al. 2011. Efficient electrochemical oxidation of perfluorooctanoate using a Ti/SnO2-Sb-Bi anode[J]. Environmental Science & Technology, 45(7): 2973-2979. |