 2020, Vol. 40
2020, Vol. 40



汞(Hg)是一种危害人类健康和生态系统的全球性重金属污染物, 具有不可降解、广泛迁移和生物富集等特性.除火山喷发等自然活动外, 矿山开采和工业活动等人为活动是造成全球范围内汞浓度增加的主要原因, 目前已受到人们的广泛关注(Krabbenhoft et al., 2013; 陈雅丽等, 2019).我国人为排放的汞污染主要来源包括有色金属冶炼和煤炭燃烧, 其余则来自水泥生产、生活垃圾焚烧等(Cheng et al., 2012), 这些不同来源的汞污染物又以不同的途径(大气沉降、固废堆置、地表径流等)进入土壤和地下水, 从而严重威胁人类健康和生态系统.
在水体环境中, 无机汞存在3种价态, 即Hg(0)、Hg(I)、Hg(II), 且在一定程度上可相互转化, 这主要取决于水环境的氧化还原电位.Hg(I)和Hg(II)分别以二聚物(Hg22+)和Hg2+的形式存在, 同时也以多种络合物形式存在, Hg22+容易发生歧化反应生成Hg2+和Hg0, 而Hg2+具有较强的水溶性, 在水环境中可以发生络合、吸附、甲基化等反应, 并通过生物和非生物化学还原转化为Hg0(Zhu et al., 2018).近年来, 大多数对汞污染的研究集中在大气、地表水和土壤污染方面(Gai et al., 2016;童银栋等, 2016; 王馨慧等, 2016), 而在地下水污染方面的研究则相对较少.如Andrea等(2018)研究了河流冲积平原浅层含水层中汞的空间分布和形态特征, 发现物理化学边界条件如pH、氧化还原电位Eh和溶解氧的变化及地下水位的动态变化会影响地下水中汞的迁移及转化.Johannesson等(2013)建立了汞在深部地下水中一维生物地球化学反应溶质运移模型, 并考虑了微生物作用下铁与硫酸盐还原作用、含铁矿物溶解及汞吸附矿物后产生共沉淀对地下水的汞浓度变化的影响.目前, 国内对汞在地下水中迁移转化行为的研究较少.研究表明, 汞污染物在地下水中的迁移、转化规律受复杂的物理、化学及生物地球化学作用的影响, 并以各种无机或有机配体的形式存在, 从而造成了不同形态汞的毒性和环境行为方面均具有显著差异(Gabriel et al., 2014).
一般而言, 直接测定汞的形态具有较大的难度.与此相比, 水文地球化学模型可作为分析汞的化学反应过程的重要工具, 进而构建场地条件下的反应性溶质运移模型.目前, 数值模型已成为将地下水溶质运移和考虑复杂生物地球化学反应相耦合的有力工具(Prommer et al., 2019), 可用来模拟和预测汞污染物在地下水中的迁移转化规律并进行风险评价.基于此, 本文以德国南部某遗留木材加工厂场地为例, 结合场地地下水中汞污染特征及监测数据, 利用PHREEQC模型研究地下水中无机汞的存在形态分布, 然后耦合水文地球化学反应与对流、弥散过程, 构建汞在地下水中迁移的多组分反应溶质运移模型.通过分析汞在该特定场地条件下的迁移、转化规律, 为定量预测其变化趋势及制定相应的修复措施提供重要的科学支撑.
2 研究区概况(Overview of research area) 2.1 污染场地概况研究区位于德国南部的一个木材加工厂旧址, 占地面积约9×104 m2.该工厂将氯化汞(HgCl2)溶液用作木材防腐剂, 在过去持续近半个世纪(1904 —1965年)的木材加工过程中, 由于不合理堆放导致约有10~20 t汞释放到土壤和含水层中.自20世纪70年代起该工厂的旧址陆续改造为居民区, 通过采用地表土壤物理挖掘迁移的方式, 已去除了污染源处表层的汞污染物.由于土壤中汞浓度较高, 渗流通量低, 大部分汞污染物仍持续渗入到含水层中.如图 1所示, E3为主要的泄漏点, 最终形成了一个长约1.3 km、宽100 m的地下水汞污染羽, 污染羽的中心浓度达到450 μg·L-1, 已经远远超过世界卫生组织(World Health Organization, WHO)规定的饮用水中汞最大浓度(1 μg·L-1)(WHO, 1993).该场地位于地下水保护区内, 土壤和地下水汞污染已对保护区地下水资源造成了严重威胁(Schöndorf et al., 1999; Bollen et al., 2008).
 |
| 图 1 场地地下水汞污染羽分布平面图(引自Bollen et al., 2008) Fig. 1 Plan view of site and groundwater Hg contamination plume(After Bollen et al., 2008) |
研究场地实际剖面图如图 2所示, 沿污染羽方向布置了6口地下水观测井(B14、P20、P19、B10、B28、B3).场地水文地质结构较为简单, 地表以下1~3 m主要为人工回填土和建筑碎石, 天然土壤埋深约5~6 m, 主要是有机碳含量较低的淤泥钙质粉土.地下水位标高约为4~10 m, 含水层岩性主要是渗透性较高的松散砾石层, 地下水水力梯度为7‰~10‰, 地下水实际流速约为3~10 m·d-1, 溶解有机碳(DOC)含量极低, 含水层底部为低渗透性风化砾岩, 构成相对隔水层.
 |
| 图 2 场地实际剖面图 Fig. 2 Profile of the study site |
地下水观测孔的水样检测结果表明, 水化学特征相对均一.各主要离子组分浓度变化范围不大, 地下水中汞的背景浓度低于检出限(<0.1 μg·L-1), pH为6.89~7.65, Eh为383~480 mV, 污染源含高浓度的氯离子.根据批实验估算出从土壤中淋滤出的汞浓度为3 ~11000 mg·kg-1, 图 2中泄露点E3地表以下2 m处淋滤出的汞浓度为11048 mg·kg-1, 随着深度的增加逐渐降低至23 mg·kg-1, 然后运移至地下水中并在对流、弥散与化学反应等过程下扩散至下游.通过分析地下水中提取的各形态汞(具体分析测试方法参见文献(Bollen et al., 2011), 将地下水中总汞定义为总溶解性汞Hgtot, 并进一步分为4种形态:溶解性金属汞(Hg0)、易还原性溶解无机汞(HgIIa, 包括HgCl2、Hg(OH)2等)、有机质结合态汞(HgIIb)、固体基质结合态汞(Hgpart).
3 汞的反应运移模型(Mercury reactive transport model) 3.1 水流与运移模型基本控制方程基于质量守恒定律的基本原则, 研究区地下水稳定流场可由水流控制方程和达西定律计算:

|
(1) |
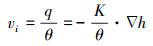
|
(2) |
假设多孔介质有Nc种组分, 每一种组分以水相或固相存在, 对于任一种组分k的总浓度Tk(M·M-1)包括水相组分浓度Ck和固相组分浓度Sk, 并满足式(3)(Walter et al., 1994).
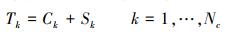
|
(3) |
任一种水相组分k在多孔介质中的运移可以用对流-弥散方程来描述:

|
(4) |
对于固相组分k, 则有:
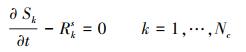
|
(5) |
式中, t为时间(T), h为水头(L), q为单位流量(L·T-1), θ为有效孔隙度, K为渗透系数(L·T-1), vi为平均孔隙水流速(L·T-1), Dij为水动力弥散系数张量(L2·T-1), 取决于纵向弥散度αL及横向弥散度αT(L)和分子扩散系数Dd(L2·T-1), Rk为化学反应源汇项(M·M-1·T-1), 仅表示水相组分浓度的变化, Rsk为溶解沉淀和吸附解吸反应引起固相组分浓度的变化(M·M-1·T-1).
3.2 反应过程与数值模拟程序PHT3D(Prommer et al., 2003)是由溶质运移模型MT3DMS(Zheng et al., 1999)与地球化学反应模型PHREEQC-2(Parkhurst et al., 1999)耦合而成的多组分化学迁移程序.由于PHREEQC-2的灵活性和广泛适用性, 因此, PHT3D能处理上述对流-弥散方程中化学反应项的一系列平衡和动力学控制的生物地球化学反应过程, 包括均相(水相)络合反应、矿物的溶解-沉淀、双电层表面络合模拟、离子交换及氧化还原等反应过程.由质量作用定律确立的部分平衡反应过程如表 1所示.
| 表 1 地球化学平衡反应质量作用方程 Table 1 Equilibrium geochemical mass-action equations |
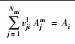
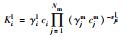
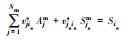
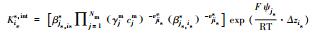
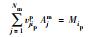
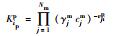
表 1中Kil、Kies, int、Kipp分别为水相络合平衡常数、考虑静电项修正的表面平衡常数及矿物溶解平衡常数, Nm为主要组分的数目, vji为化学计量系数, Ajm、Ai、Mip分别为水相主要组分、次要组分和矿物化学表达式, cj、ci分别为j种主要组分浓度与i种次要组分浓度(mol·kg-1), γjm、γil分别为主要组分和次要组分活度系数(kg·mol-1), F为法拉第常数, ψjs为吸附表面电位(V), zis为离子电荷, R为理想气体常数(kJ·mol-1·K-1), T为绝对温度(K).
3.3 数值模型首先利用地下水水流模拟程序MODFLOW-2005(Harbaugh, 2005)建立二维剖面上的地下水稳定流模型.模拟区域水平距离为1600 m, 垂直厚度为10 m, 采用均匀网格将模拟区域离散为101列和40层.水流模型的范围及边界条件见图 3, 忽略降水入渗补给及地下水水位波动的影响, 左右两侧按定水头边界处理, 水位分别保持在4 m和10 m左右, 含水层底部概化为零流量边界.反应运移模型与水流模型的范围相同, 潜水面为Cauchy边界, 边界通量为污染物泄露速率0.0022 m·d-1, 其他边界视为Neumann边界条件, 穿过这些边界的浓度梯度为零.研究区域含水层主要参数设置为:水平渗透系数为3×10-3 m·s-1, 垂直与水平渗透系数之比为0.1, 孔隙度为0.25, 纵向弥散度为10 m, 横纵弥散度比为0.1, 模拟应力期为600 d, 划分为60个时间步长.基于MODFLOW-2005计算的流场, 使用MODPATH程序进行粒子追踪, 得出溶质在地下水中的运动轨迹, 即粒子迹线如图 3所示.
 |
| 图 3 概念模型及边界条件 Fig. 3 The conceptual model and boundary conditions |
基于水流模型, 采用PHT3D程序模拟污染源处氯化汞溶液以浓度C0持续泄露地下水中的对流-弥散-反应过程.模型中的组分包括Na+、Ca2+、Cl-、Br-、O2(aq)、DOC、Hg2+、Hg0等, 采用Davies方程计算各组分的活度系数.反应过程主要考虑汞与无机配体及溶解性有机质的络合反应、与无定形态水合铁矿的表面络合吸附反应及氧化还原反应, 不包括汞的甲基化过程和金属汞的挥发过程, 假设部分反应处于热力学平衡状态.如表 2所示, 模型中化学反应体系包含10个水相络合反应与3个与有机质络合反应, 汞与无机配体通过络合反应生成各种络合物, 且与有机质能形成稳定的络合物, 其中, 硫醇基团(RS)是有机质与汞结合强的官能团(张孟孟等, 2011), 络合反应平衡常数取自文献(Skyllberg, 2008), 硫醇基团(RS)与溶解有机碳(DOC)之比取Richard等(2016b)拟合出的最佳质量分数为0.03%.模型中通过电荷平衡后的主要组分初始浓度及污染源处浓度如表 3所示, 且含水层的主要矿物石英、方解石和非晶态Fe(OH)3参与溶解-沉淀反应.
| 表 2 汞与部分无机配体及溶解性有机质络合反应平衡常数 Table 2 Aqueous complexation reactions equilibrium constants for Hg with inorganic ligands and dissolved organic matter |
| 表 3 反应性溶质运移模型的初始及边界组分浓度 Table 3 Initial and boundary component concentrations for reactive transport simulation |
吸附是控制汞在地下水中迁移的主要机制, 传统上吸附反应基于经验的等温吸附模型, 如线性等温吸附(Kd)、Langmuir等温吸附与Freundlich等温吸附.等温吸附模型在描述溶质在液相和固相之间的分配虽简单易用, 但不能反映溶质的吸附程度随水化学条件及pH值的时空变化.汞的组分及浓度、水化学条件、矿物表面的交替变化及有机物的影响, 在时间和空间上都不能假定汞在迁移过程存在恒定的阻滞因子(Schöndorf et al., 1999).鉴于此, 以双电层理论为基础的表面络合模型(Surface Complexation Model, SCM)更具有理论依据, 能很好地描述固-液界面上发生的化学反应.在大多数含水层中, 水合氧化铁(HFO)是非常重要的微量金属吸附剂, 对汞在迁移中的吸附阻滞作用已有部分研究(Richard et al., 2016a).本文采用双电层表面络合模型模拟二价汞吸附到含水层中无定形态水合氧化铁矿物(HFO)表面.HFO主要来源于非晶态Fe(OH)3的沉淀, 只考虑一个吸附位点SurfaOH, 包括HFO表面吸附位点与Hg(Ⅱ)、OH-和Cl-之间反应的三元表面络合物(Tiffreau et al., 1995).表面络合反应及对应的平衡常数见表 4.其中, 吸附剂的表面位点密度与Fe(OH)3物质的量有关, 假定每摩尔Fe(OH)3表面含吸附位点0.015 mol.
| 表 4 反应体系中表面络合反应平衡常数logK (298.15 K, I=0) Table 4 Surface complexation constants in the reaction network |
氧化还原作用是影响汞在地下水中存在形态、迁移转化的重要因素.在一定程度上, 二价铁(Lamborg et al., 2013)、溶解性有机质(Gu et al., 2011)及微生物(Wiatrowski et al., 2006)均可作为电子供体将二价汞还原为金属汞, 还原生成的溶解金属汞Hgaq0较容易挥发至大气中, 那么水相Hg2+转化为气相Hg0涉及两个反应(Leterme et al., 2014), 即还原反应(6)和挥发反应(7).
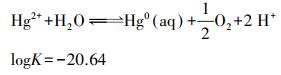
|
(6) |
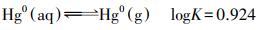
|
(7) |
研究表明, 厌氧环境中含铁矿物可作为电子受体, 其动力学还原过程对溶解金属汞Hgaq0的形成起着重要作用.铁主要以氧化铁矿物磁铁矿(Fe3O4)的形式在沉积物中富集, 其还原动力过程可以用一级动力学模型来描述:
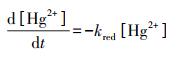
|
(8) |
式中, [Hg2+]为反应体系中二价汞的浓度(mol·L-1), kred为Hg2+反应速率常数(mol·m-2·s-1).在实验条件下, 二价汞的还原反应速率受磁铁矿表面积、pH值和Cl-浓度的影响(Wiatrowski et al., 2009), 本次模拟取与场地水化学条件较相似的反应速率常数(5.0×10-4 mol·m-2·s-1).
4 结果与讨论(Results and discussion) 4.1 无机二价汞的存在形态利用地球化学模拟程序PHREEQC模拟场地无机二价汞Hg(II)的各种化学存在形态, 地下水水样数据来自文献(Richard et al., 2016b), 并分别采用数据库minteq.v4.dat和数据库Thermoddem(Blanc et al., 2012), 两者都未包含汞与有机质络合反应的平衡常数.针对两种数据库采用PHREEQC程序计算出的各无机汞形态含量对比见图 4, 可以看出无机汞的形态非常相似, 只有部分形态存在较小差异, 这主要是由于数据库包含汞的形态和平衡常数不同, 如分别存在HgBrCl与HgCl3-.采用不同数据库模拟出的水样中无机化合态汞均以HgCl2为主要存在形态, 含量分别达53.17%~57.03%、72.23%~76.06%, 其次为HgClOH, 含量分别约为19%、33%.此外, Hg(OH)2也占有一定比例, 水样中其它形态含微量的HgBrOH、HgBr2与HgCl+等.pH值、离子强度及氧化还原电位是影响汞在水体中存在形态的关键因素, 具有较强亲和力的配体如Cl-、OH-与Hg2+形成的络合物, 可以大大提高汞的溶解度, 使汞在水中的迁移能力提高.因此, 地下水中存在的无机化合态汞以HgCl2、HgClOH含量较高, 并因地下水水化学条件不同而异.综合考虑数据库minteq.v4.dat和数据库Thermoddem所包含的汞形态, 并通过添加其它化学反应及其相关的平衡常数, 将作为下一步反应运移模型的热力学数据库.
 |
| 图 4 无机二价汞的存在形态模拟与对比 Fig. 4 Simulation and comparison of divalent inorganic mercury speciation |
综合考虑汞在地下水迁移中的对流-弥散物理过程与离子络合反应、表面络合吸附及氧化还原反应地球化学过程, 图 5分别展示了反应运移模型模拟出的二价汞与金属汞运移100、250、600 d后的污染羽分布范围.由于地下水流速较快, 运移主要以对流运动为主, 弥散作用与之相比可以忽略, 污染羽在较短的时间内很快地运移至较远距离, 经计算从污染源运移至左边界约需350 d.从图中可以看出, 二价汞的污染羽范围不断扩大, 浓度最低值大于地下水背景值浓度(< 0.1 μg·L-1);金属汞由于二价汞的还原作用, 中心浓度随污染羽的范围不断变化, 其中心浓度值可达到5.07×10-8 mol·L-1.虽然本文建立的反应运移模型缺乏详细的场地数据进行校准, 但仍能对汞污染羽的迁移转化过程进行定量描述, 再现污染羽浓度的总体特征.图 6为观测孔P19及观测孔B14处汞吸附至水合铁表面各种形态及pH随时间变化的模拟结果, 3种不同吸附形态的汞随时间变化趋势一致, 但各自的浓度存在差异.表面络合模型反映出运移过程中吸附能力随水化学条件的变化, 将不同吸附形态汞的浓度相加计算得到吸附汞的总量, 相对于水铁矿的质量分数分别仅为0.346%和0.0176%, 而且相比溶解性汞的浓度值小2~3个数量级, 说明在流速较大的地下水系统中表面络合吸附作用对汞的迁移阻滞作用较小.
 |
| 图 5 反应运移模拟污染羽的时空演化 Fig. 5 The evolution of contamination plume using reactive transport modeling |
 |
| 图 6 汞吸附形态与pH变化模拟结果(a.观测井P19, b.观测井B14) Fig. 6 Results of mercury sorbed specitation and pH at well locations P19 and B14 |
图 7为反应运移过程中各观测井测量出的各形态汞占总汞的比例与模拟结果的对比, 图中“实测1”分析数据来自文献Bollen等(2008), “实测2”分析数据来自文献Richard等(2016b).通过与模拟值对比, 可以明显看出无机汞HgIIa占主导, 且主要以溶解态HgCl2的形式存在, 最初也是以此形态释放到土壤与地下水中.测量结果表明, 溶解金属汞Hgaq0占总汞的比例仅为0.7%±0.4%, 而模拟出的比例达到3.2%~12.6%.模拟出的溶解金属汞Hgaq0主要来源于无机汞的还原作用.Richard等(2016a)认为在实际场地中金属汞Hgaq0一方面相比HgIIa容易挥发, 其亨利常数达1.1×10-1 mol·L-1·atm-1, 而无机汞HgIIa的亨利常数为1.4×10-6 mol·L-1·atm-1, 土壤中析出气相Hg0也佐证了这一点;另一方面也易吸附到水合氧化铁(HFO)表面并发生共沉淀;在这两方面的共同作用下导致测量与模拟出的金属汞Hgaq0含量存在较大差异.运移过程有机质结合态汞HgIIb占据了一部分比例(0.9%~6.0%), 虽然地下水中有机质含量较少, 但汞与有机质具有较强的结合能力, 主要通过—S和—O等官能团与有机质(DOM)发生强烈的结合.最后与测量出的汞吸附到含水层固体基质表面的量相比, 由于固体基质包括石英砂和水铁矿等矿物, 而模拟主要考虑汞吸附到水合铁矿物表面, 因此, “实测1”测量出的固体基质结合态汞含量相比“实测2”测量值与模拟出的值较大, 总体而言其含量可忽略不计.不同形态汞的迁移转化模拟结果进一步表明在迁移过程中汞大部分仍以溶解性二价汞形式存在, 二价汞还原为金属汞为主要转化过程, 但零价汞的挥发作用不容忽视.另外, 即使地下水中有机质(DOM)浓度较低的情况下, 当Hg-DOM结合位点比例较高时, 汞与有机质通过络合作用形成的Hg-DOM络合物通常也占据为主要形态.
 |
| 图 7 4种不同形态汞所占比例模拟与实测值(O1:实测1;O2:实测2;S:模拟值) Fig. 7 Simulated(S) and observed(O1, O2) proportion of four different species of mercury |
1) 采用地球化学模拟程序PHREEQC并通过对比两种热力学数据库, 分析了污染场地地下水中无机二价汞Hg(II)的主要存在形态, 结果显示, 形态HgCl2与HgClOH占优势, 主要原因在于地下水中Cl-浓度较高, 同时Cl-与Hg2+具有较强的亲和力, 形成的络合物溶解度较高, 可以大大增加汞的迁移性.
2) 汞在场地地下水迁移转化过程中的反应运移模型揭示了由动力学控制的二价汞转化为金属汞的氧化还原作用是影响汞在地下水迁移转化的一个重要因素.此外, 不同表面吸附形态汞的浓度与pH的变化趋势, 表明HFO对汞的迁移阻滞没有显著影响.
3) 在迁移转化过程中汞的形态分布特征存在明显的差异, 其主要形态大部分以溶解性二价汞存在, 一部分发生转化过程, 如还原为金属汞, 与有机质结合形成Hg-DOM络合物, 同时可以发现几乎不含有水铁矿结合态汞.由此可作为原位修复汞污染地下水的参考依据, 如可渗透反应墙(PRB)内填充一些反应活性吸附剂, 通过电子转移使汞还原为金属汞, 最后与吸附剂发生共沉淀达到较好的修复效果.
Andrea C, Stefano C, Andrea E, et al. 2018. Mercury in the unconfined aquifer of the Isonzo/Soča River alluvial plain downstream from the Idrija mining area[J]. Chemosphere, 195: 749-761. DOI:10.1016/j.chemosphere.2017.12.105 |
Blanc P, Lassin A, Piantone P, et al. 2012. Thermoddem:A geochemical database focused on low temperature water/rock interactions and waste materials[J]. Applied Geochemistry, 27(10): 2107-2116. DOI:10.1016/j.apgeochem.2012.06.002 |
Bollen A, Biester H. 2011. Mercury extraction from contaminated soils byl-cysteine:Species dependency and transformation processes[J]. Water Air and Soil Pollution, 219(1/4): 175-189. |
Bollen A, Wenke A, Biester H. 2008. Mercury speciation analyses in HgCl2-contaminated soils and groundwater-Implications for risk assessment and remediation strategies[J]. Water Research, 42(1/2): 91-100. |
Cheng H, Hu Y. 2012. Mercury in municipal solid waste in China and its control:A review[J]. Environmental Science & Technology, 46(2): 593-605. |
陈雅丽, 翁莉萍, 马杰, 等. 2019. 近十年中国土壤重金属污染源解析研究进展[J]. 农业环境科学学报, 38(10): 2219-2238. DOI:10.11654/jaes.2018-1449 |
Dzombak D A, Morel F M M. 1990. Surface Complexation Modeling:Hydrous Ferric Oxide[M]. New York: Wiley.
|
Gabriel M C, Williamson D G. 2014. Principal biogeochemical factors affecting the speciation and transport of mercury through the terrestrial environment[J]. Environmental Geochemistry and Health, 26(3/4): 421-434. |
Gai K, Hoelen T P, Hsu-Kim H, et al. 2016. Mobility of four common mercury species in model and natural unsaturated soils[J]. Environmental Science & Technology, 50(7): 3342-3351. |
Gu B, Bian Y, Miller C L, et al. 2011. Mercury reduction and complexation by natural organic matter in anoxic environments[J]. Proceedings of the National Academy of Sciences of the United States of America, 108(4): 1479-1483. DOI:10.1073/pnas.1008747108 |
Harbaugh A W.2005.MODFLOW-2005, The U.S.Geological Survey Modular Ground-Water Model-the Ground-Water Flow Process[R].Reston: US Geological Survey https://pubs.er.usgs.gov/publication/tm6A16
|
Johannesson K H, Neumann K. 2013. Geochemical cycling of mercury in a deep, confined aquifer:Insights from biogeochemical reactive transport modeling[J]. Geochimica et Cosmochimica Acta, 106: 25-43. DOI:10.1016/j.gca.2012.12.010 |
Krabbenhoft D P, Sunderland E M. 2013. Global change and mercury[J]. Science, 341(6153): 1457-1458. DOI:10.1126/science.1242838 |
Lamborg C H, Kent D B, Swarr G J, et al. 2013. Mercury speciation and mobilization in a wastewater-contaminated groundwater plume[J]. Environmental Science & Technology, 47(23): 13239-13249. |
Leterme B, Blanc P, Jacques D. 2014. A reactive transport model for mercury fate in soil-Application to different anthropogenic pollution sources[J]. Environmental Science and Pollution Research, 21(21): 12279-12293. DOI:10.1007/s11356-014-3135-x |
Parkhurst D L, Appelo C A.1999.User's guide to PHREEQC (Version 2): A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations[R].Denver: Geological Survey
|
Prommer H, Barry D A, Zheng C. 2003. MODFLOW/MT3DMS-based reactive multicomponent transport modeling[J]. Groundwater, 41(2): 247-257. DOI:10.1111/j.1745-6584.2003.tb02588.x |
Prommer H, Sun J, Kocar B D. 2019. Using reactive transport models to quantify and predict groundwater quality[J]. Elements:An International Magazine of Mineralogy, Geochemistry, and Petrology, 15(2): 87-92. |
Powell K J, Brown P L, Byrne R H, et al. 2004. Chemical speciation of Hg(II) with environmental inorganic ligands[J]. Australian Journal of Chemistry, 57(10): 993. DOI:10.1071/CH04063 |
Richard J H, Bischoff C, Ahrens C G M, et al. 2016a. Mercury(II) reduction and co-precipitation of metallic mercury on hydrous ferric oxide in contaminated groundwater[J]. Science of the Total Environment, 539: 36-44. DOI:10.1016/j.scitotenv.2015.08.116 |
Richard J H, Bischoff C, Biester H. 2016b. Comparing modeled and measured mercury speciation in contaminated groundwater:Importance of dissolved organic matter composition[J]. Environmental Science & Technology, 50(14): 7508-7516. |
Schöndorf T, Egli M, Biester H, et al. 1999. Distribution, Bioavailability and Speciation of Mercury in Contaminated Soil and Groundwater of a Former Wood Impregnation Plant//Mercury Contaminated Sites[M]. Berlin: Heidelberg:Springer.
|
Skyllberg U. 2008. Competition among thiols and inorganic sulfides and polysulfides for Hg and MeHg in wetland soils and sediments under suboxic conditions:Illumination of controversies and implications for MeHg net production[J]. Journal of Geophysical Research:Biogeosciences, 113: G00C03. |
Tiffreau C, Lützenkirchen J, Behra P. 1995. Modeling the adsorption of mercury (II) on (hydr) oxides:I.Amorphous iron oxide and α-quartz[J]. Journal of Colloid and Interface Science, 172(1): 82-93. DOI:10.1006/jcis.1995.1228 |
童银栋, 张巍, 邓春燕, 等. 2016. 大气汞均相和非均相化学反应过程研究进展[J]. 环境科学学报, 36(5): 1515-1523. |
Walter A L, Frind E O, Blowes D W, et al. 1994. Modeling of multicomponent reactive transport in groundwater:1.Model development and evaluation[J]. Water Resources Research, 30(11): 3137-3148. DOI:10.1029/94WR00955 |
Wiatrowski H A, Das S, Kukkadapu R, et al. 2009. Reduction of Hg(II) to Hg(0) by magnetite[J]. Environmental Science & Technology, 43(14): 5307-5313. |
Wiatrowski H A, Ward P M, Barkay T. 2006. Novel reduction of mercury (II) by mercury-sensitive dissimilatory metal reducing bacteria[J]. Environmental Science & Technology, 40(21): 6690-6696. |
World Health Organization.1993.Guidelines for Drinking Water Quality[S].Geneva, Switzerland: WHO
|
王馨慧, 单保庆, 唐文忠, 等. 2016. 典型城市河流表层沉积物中汞污染特征与生态风险[J]. 环境科学学报, 36(4): 1153-1159. |
Zheng C M, Wang P P.1999.MT3DMS: A Modular Three-Dimensional Multispecies Transport Model for Simulation of Advection, Dispersion, and Chemical Reactions of Contaminants in Groundwater Systems; Documentation and User's Guide[R].Tuscaloosa: Alabama University
|
Zhu S, Zhang Z, Žagar D. 2018. Mercury transport and fate models in aquatic systems:A review and synthesis[J]. Science of the Total Environment, 639: 538-549. DOI:10.1016/j.scitotenv.2018.04.397 |
张孟孟, 戴九兰, 王仁卿. 2011. 溶解性有机质对土壤中汞吸附迁移及生物有效性影响的研究进展[J]. 环境污染与防治, 33(5): 95-99+110. DOI:10.3969/j.issn.1001-3865.2011.05.021 |