 2020, Vol. 40
2020, Vol. 40



大气气溶胶是“痕量元素及其同位素的海洋生物地球化学循环研究”国际海洋科学计划(GEOTRACES)的研究内容之一, 目前已受到广泛重视.气溶胶可通过干沉降(灰尘、土壤、灰分)和湿沉降(雨、雾、雪)输入到表层海洋, 是浮游植物微量营养元素和有毒重金属的重要来源(Shelley et al., 2017), 同时会影响浮游植物群落结构和生态系统(Bishop, 2002).除了研究最多的Fe会对浮游植物生长产生限制之外, 一些报道指出Zn、Co和Mn也是限制浮游植物生长的微量元素(Saito et al., 2008;Browning et al., 2014), 一些微量元素如Cu在较高浓度时会对浮游植物产生毒害作用(Mahowald et al., 2018).黄海海域是受亚洲沙尘影响最大的近海海域之一(张凯等, 2005), 2007年两次大规模亚洲沙尘暴期间, 该海域溶解态铁、磷和总无机氮的输送通量约是非沙尘暴期间的15、13和5倍, 沙尘暴过后3~4 d黄海爆发水华(Shi et al., 2012; Shi et al., 2013).
气溶胶中某些微量金属元素(如Al、Fe、Ti和Mn)主要受沙尘来源控制(Doney et al., 2009), 人类活动和火山喷发也是气溶胶中微量金属元素(如Pb、Zn和Cd)的重要来源(Mahowald et al., 2018).金属溶解度受元素本身性质(Chester et al., 1993)及其在颗粒物中存在形式的影响(Qureshi et al., 2006), 同时气溶胶中微量元素来源对其溶解度影响也较大(Hsu et al., 2010), 并且不同区域、季节气溶胶中微量元素的溶解度差异较大, Mahowald等(2005)指出气溶胶中Fe的溶解度变化范围可达0.01%~80%.
大气输送的微量元素在海洋表层达到溶解平衡和移除时间能够反映气溶胶生物化学过程的许多信息, 如微量元素溶解和移除过程, 以及生物可利用程度等(Bridgestock et al., 2016), 但目前关于这方面的研究很少(Kadko et al., 2019).为了获得气溶胶中微量元素在海洋表层达到溶出平衡的相关信息, 最直接的方法是将气溶胶样品用海水淋溶提取, 但在实际操作中却极为困难(Baker et al., 2010).Eyckmans等(2001)设计的微循环淋溶技术可以研究金属溶出的动力学过程, 用于海洋表层气溶胶微量元素溶出行为的研究.
基于此, 本文以夏季南海为研究对象, 测定气溶胶中微量元素总浓度(CT)及溶解态浓度(CS), 计算其溶解度(s)并估算微量元素干沉降通量;同时, 运用后向轨迹分析、富集因子等方法对元素进行源解析, 并通过溶解动力学实验, 深入探究影响气溶胶微量元素溶解度及溶出过程的因素, 以初步探讨大气沉降对南海东北部初级生产力的影响.
2 材料与方法(Materials and methods) 2.1 样品采集于2017年6月9日—8月11日搭载“东方红2”科考船“南海东北部-吕宋海峡共享航次(NORC2017-05)”, 采集南海东北部10个气溶胶样品, 采样期间盛行西南季风, 避开了热带低压和台风活跃期.采样过程航迹如图 1所示, 其中, 6月21日和7月12日采样期间有降雨事件发生.
 |
| 图 1 南海气溶胶采样航迹图 Fig. 1 The cruise track with aerosol sample deployment locations in South China Sea |
将TSP采样器(崂应2031型)放置于“东方红2”船最高甲板前端采集气溶胶样品.为防止海面飞沫、轮船煤烟和湿沉降带来的影响, 当相对风速小于0.5 m · s-1、相对风向超过±60°或下雨时停止采样.采样滤膜为GEOTRACES推荐的Whatman 41纤维素膜, 有效采样面积为230 mm×180 mm.使用前用pH=2的盐酸浸泡滤膜24 h, 然后用Milli-Q水洗至中性, 45 ℃烘干称量初重.在简易洁净工作台中将清洗过的采样膜固定在滤膜夹上, 放入洁净塑封袋内, 带到室外采样器放入采样夹内固定.采样流量设置为1.05 m3 · min-1, 采样时间设置为20 h(特殊情况因天气不佳不足20 h), 样品标准体积为550~2000 m3, 实际采样时长和体积见表 1, 因测定含量为相对量, 所以不同采样体积对后续试验没有影响.采样结束后, 于简易洁净工作台上将样品膜对折后装入洁净塑封袋冷冻保存.在岸基实验室内, 在45 ℃下烘干滤膜并称量末重.采样膜末重与初重差值为所采集颗粒物的质量.
| 表 1 采样日期、时长和体积 Table 1 Sampling date, duration, and volume |
实验所使用的Merck HNO3、HCl均在洁净实验室经过亚沸蒸馏提纯, 以降低空白值.本研究采用微循环淋溶法(Eyckmans et al., 2001), 微循环淋溶系统(图 2)在使用前用pH=2的盐酸溶液冲洗20 min, 之后用Milli-Q水冲洗至中性.
 |
| 图 2 微循环淋溶装置 Fig. 2 The mini leaching systems for solubility experiments |
气溶胶中微量元素溶解态浓度(CS)测定:将采样膜用洁净陶瓷剪刀裁剪为30 mm×60 mm大小的膜, 在微循环淋溶模式下, 用10 mL Milli-Q水, 以2 mL · min-1的流速循环淋溶45 min, 保留淋溶液待测.
气溶胶中微量元素的溶解动力学研究:将采样膜用洁净陶瓷剪刀裁剪为30 mm×60 mm大小的膜, 系统调为连续淋溶模式.前55 min用Milli-Q水作淋溶液, 55 min后用pH=2的HCl溶液作淋溶液, 流速均为2 mL · min-1.每阶段前20 min每2 min、后35 min每5 min从出液口接取淋溶液待测.
气溶胶中微量元素总含量(CT)测定:选取5个能够代表气团来源的样品(6月14、21日, 7月9、13、20日)进行微量元素总含量测定.用洁净的陶瓷剪刀截取60 mm×60 mm的样品膜, 放入聚四氟乙烯消化罐内, 消化罐使用前用30%硝酸溶液浸泡24 h.加入3 mL浓HNO3、3 mL HClO4、1 mL HF密封后用加热板180 ℃加热12 h, 然后开盖180 ℃加热赶酸至近干, 冷却后加入3 mL浓HNO3、1 mL HF密封后用加热板180 ℃加热12 h, 然后开盖180 ℃加热赶酸至近干, 冷却后用2% HNO3定容待测.
样品经预处理后, 以Rh为内标元素用ICP-MS(TC017-iCAPQ -NexION300X, 美国赛默飞世尔科技-珀金埃尔默公司)测定Al、Fe、Mn、Cu、Zn、Cr、Co、Ni、Pb、Ti、V、Ba的浓度, 检出限Al为0.061 μg · L-1, Fe为0.071 μg · L-1, Mn为0.082 μg · L-1, 其它元素均低于0.04 μg · L-1.从一张采样膜上裁剪下5张相同大小的膜, 进行平行试验检测精密度, 各元素的相对标准偏差均小于8%.将国家水系沉积物标准GSD-9进行消解(n=5)分析, 回收率为100%~106%.
2.3 溶解度、EF值和后向轨迹分析气溶胶中元素溶解度(s)、富集因子(EF)及气溶胶中元素累积溶解度(st)的计算公式分别如式(1)~(3)所示.
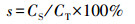
|
(1) |

|
(2) |
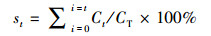
|
(3) |
式中, CS为气溶胶中元素溶解态浓度(ng · m-3), CT为气溶胶中元素总浓度(ng · m-3), (Cx/CAl)aerosol为气溶胶样品中某元素与Al浓度的比值, (Cx/CAl)crust为地壳中该元素与Al浓度的比值(Taylor, 1964), t为淋溶时间(min), Ct为各个时间段元素浓度(ng · m-3).
后向轨迹分析:利用美国海洋与大气局(NOAA)空气资源实验室(ARL)和澳大利亚气象研究中心联合开发的混合型单粒子拉格朗日综合轨迹模式(HYSPLIT_4, Hybrid Single-Particle Lagrangian Integrated Trajectory, http://www.arl.noaa.gov/)进行后向轨迹分析.后向轨迹起始时间选择气溶胶样品采集的起始时间, 起始高度选择500 m, 向前48 h得到气团运动轨迹.
3 结果与讨论(Results and discussions) 3.1 夏季南海东北部大气气溶胶微量元素浓度和溶解度夏季南海东北部气溶胶颗粒物浓度为1.6~49.9 μg · m-3, 平均值为(24.2±15.8) μg · m-3, 与吴兑(1995)在南海北部海岛测得的浓度(23.4 μg · m-3, 1998—1990年两年均值)接近, 远小于东海(62.8 μg · m-3, 2006—2008春、夏、冬季均值)(石金辉等, 2011)和黄海(108.3 μg · m-3, 2011年春季)(Zhao et al., 2015)的浓度值.6月21日和7月12日样品采集过程中存在降水, 雨水冲刷使空气中大气颗粒物浓度降低, 分别为3.5 μg · m-3和1.6 μg · m-3, 显著低于其它样品(t-检验, p < 0.01).根据后向轨迹(图 3)将样品分为3类:6月14、15、21和26日的气溶胶主要来自于南海南部, 7月9、12、13和20日的气溶胶来自于菲律宾附近西太平洋, 而8月3日的气溶胶来自于东印度洋并穿过中南半岛, 来源于南海南部、菲律宾附近西太平洋和东印度洋的气溶胶浓度分别为(23.0±11.9)、(31.9±11.1)和49.9 μg · m-3.
 |
| 图 3 南海气溶胶后向轨迹分析(500 m, 48 h) Fig. 3 Air mass back-trajectories from starting deployment locations in the South China Sea (500 m, 48 h) |
表 2给出了夏季南海东北部气溶胶中微量元素溶解态浓度、总浓度和溶解度数据.气溶胶中Al、Fe的总浓度远高于其他元素, 且变化范围较大, 二者之和约占颗粒物浓度的0.6%±0.3%, 远小于东海(3.1%)(吴天, 2010), 且Al、Fe浓度与颗粒物浓度之间均无显著相关关系(图未给出, r < 0.35, t-检验, p < 0.05).南海气溶胶中Fe总浓度((65.8±68.4) ng · m-3)远高于中部太平洋(9.1 ng · m-3, 2004—2006年两年均值)(Buck et al., 2013)、近北美北冰洋(0.077 ng · m-3, 2015年9—10月)(Kadko et al., 2019), 而小于东海(430 ng · m-3, 2005—2007年均值)(Hsu et al., 2010)、黄海(2384 ng · m-3, 2011年3—4月)(Zhao et al., 2015)和渤海(13821 ng · m-3, 2006—2007年)(姬洪亮等, 2011).
| 表 2 夏季南海东北部气溶胶微量元素溶解态浓度、总浓度和溶解度 Table 2 Dissolvable concentration, total concentration and solubility of trace elements in the aerosol samples collected in the South China Sea in summer |
来自南海南部气团的气溶胶中Al、Fe、Pb总浓度分别为(110.6±98.1)、(93.1±88.8)和(0.8±0.6) ng · m-3, 源于菲律宾附近西太平洋气溶胶中Al、Fe、Pb总浓度分别为(44.4±24.7)、(30.5±7.1)和(0.5±0.2) ng · m-3.气溶胶在海上输运过程中会结合海盐粒子从而改变气溶胶颗粒物质量及元素组成(石金辉等, 2011), 进而导致西太平洋气溶胶浓度比南海高, 但微量元素总浓度并不高.
南海气溶胶中Al、Ti、Fe的溶解度均小于10%, Pb、Cu、Zn、Ni、V、Co的溶解度均大于30%, Mn的溶解度则高达67.4%(表 2).不同元素溶解度差异较大, 这与元素本身性质密切相关.常燕等(2015)指出金属溶解度与其离子势有一定的负相关性.此外, 金属在颗粒物中的存在形式也显著影响其溶解度(Qureshi et al., 2006).Al、Fe大多数与硅酸盐结合, 可交换溶出量只占10%, 溶解度较小, 而Mn可交换溶出量可达20%~40%, 故其溶解度相对较高(Chester et al., 1993).提取介质的差别也是导致溶解度不同的重要因素(Baker et al., 2010), 表 3给出了不同研究区域气溶胶中微量元素在不同提取剂下的溶解度.以Milli-Q水为提取剂时, 南海气溶胶中的Fe溶解度显著高于北冰洋.所对比的北冰洋区域靠近北美和西伯利亚, 人口稀少, 人类活动比南海附近少, Fe多以硅酸盐结合形式存在, 溶解度较小.同一区域不同季节气溶胶微量元素溶解度差异也较大, 如Hsu等(2010)在对东海气溶胶的研究中发现, Al、Fe在冬季的溶解度分别为4%和5.5%, 而夏季则分别为9%和11%.
| 表 3 不同海域气溶胶痕量元素溶解度 Table 3 Solubility of aerosol trace elements in different sea areas |
气溶胶的来源(或原始组成)是控制元素溶解度的重要因素(Li et al., 2017).表 4给出了以Al作为地壳源参比元素得到的夏季南海气溶胶中微量元素相对于地壳的富集因子(EF).由表可知, Fe、Mn、Ti、Co的EF均小于2, 说明这些元素主要来源于地壳(Chen et al., 2003), 人类活动添加的量非常少, 特别是Al与Fe、Mn的总浓度之间和溶解态浓度之间均有显著的相关关系(r在0.92~0.98之间, p < 0.01).Ba、Cr的EF在2~10之间, 有一定程度的富集, 为人类活动和地壳混合源;Cu、Zn、Pb、V、Ni的EF均大于10, 存在较大程度的富集, 主要通过汽车尾气、工业冶炼、生活垃圾燃烧等人类活动排放进入大气中, V可能来自船舶重油燃烧(Sholkovitz et al., 2009).8月3日采集的气溶胶样品横穿人类活动较多的中南半岛(图 3), 而该样品中EF值较大的Cu、Zn、Pb元素溶解态浓度较其他样品有较为明显的增加, 说明人类活动影响显著.
| 表 4 夏季南海气溶胶中微量元素EF值 Table 4 The enrichment factor (EF) values of trace elements in aerosols samples collected in the South China Sea in summer |
为了更好地评估元素来源对溶解度的影响, 图 4给出了气溶胶微量元素EF值与溶解度(s)之间的关系, 可以看出, EF < 10的元素一般s≤10%, 地壳源的元素主要存在于矿物晶格, 溶解度相对较低.Mn、Co比较例外, 其EF值较小, 但溶解度较高.Chester等(1993)强调, 气溶胶元素的溶解度与EF值并非直接相关.Mn虽然主要来自于地壳, 但运输过程中由于受海洋上空较高含量Cl-络合的影响, 其溶解度增加(Deutsch et al., 1997);当Mn从铝硅酸盐矿物内部被Cl-络合移出时, Co也会被带到气溶胶颗粒表面, 使得质子碰撞替换更加容易, 从而导致地壳源元素Co溶解度也增加(Wayne et al., 2006).
 |
| 图 4 气溶胶中微量元素溶解度与地壳EF值(对数值)的关系 Fig. 4 Relationship between solubility of trace elements in aerosol samples and their EF value (logarithmic scale) |
此外, 存在其它因素影响气溶胶中微量元素溶解度.在气溶胶运输过程中, 气溶胶浓度及粒径随时间/距离呈指数级下降(Meskhidze et al., 2005).李鹏志等(2018)指出微量元素溶解度分布呈现细粒子高于粗粒子;Jickells等(2016)指出Fe溶解度与沙尘浓度呈现负相关.另外, 气溶胶体系由众多颗粒物组成, 比表面积大, 酸性气体(SO2、NO2和HNO3等)在表面酸化并与之反应, 进而促进金属的溶解(Meskhidze et al., 2005).同时, 气溶胶颗粒物可以作为云凝结核, 附着在颗粒物表面的水分进一步吸收大气中的酸性气体, 降低云水体系pH值(Kymar et al., 2011).颗粒物成云后经太阳光照射, 表面水分蒸发, 留下水膜的pH进一步降低(Seinfeld et al., 2006), 从而促进气溶胶元素溶解度提高.
3.3 南海气溶胶中微量元素溶解动力学本研究依次用Milli-Q水和pH=2的HCl溶液进行了南海气溶胶样品溶解动力学实验.图 5给出了实验中微量元素累积溶解度(st)与淋溶时间的关系.在用Milli-Q水作提取剂时, 初始阶段各金属的溶出速率很高, 随时间延长溶出速率减小, 25 min时达到溶出的动态平衡.在用HCl进行提取时, 大多数金属溶解度的变化趋势与Milli-Q水提取相同.Al、Fe、Ti和Ba在水相中达到动态平衡所需的时间比在酸相中少, 这与常燕等(2015)研究的上海气溶胶溶解动力学特征类似.大多数元素酸相溶出量占水相溶出量的30%~50%, 而Ba、Pb酸相溶出量是水相溶出量的1.3和1.8倍, 与常燕等(2015)报道的上海气溶胶Pb、Al、Fe酸溶出量能达水溶出量的30、3和4.5倍的结果差异较大.这可能是因为上海气溶胶的研究结果为2007—2009年两年均值, 其来源于陆源矿物碎屑的比例较高, 可以在酸相中持续溶出, 而南海气溶胶主要来源于海上, 在水相中溶出较为充分, 故在酸相中溶出较少, 从而导致二者酸溶出量较水溶出量的增量存在较大差异.
 |
| 图 5 南海气溶胶中痕量元素累积溶解度(st)动力学曲线 Fig. 5 Kinetic dissolution curves of cumulative solubility (st) of trace elements in the aerosol samples in the South China Sea |
水相中溶解度(pH=5~6)代表大气颗粒物中金属可溶程度的下限, 这部分金属易被生物利用, 存在强的环境迁移性;而“水+酸”溶解度(pH=2)代表可溶金属的上限, 它表示金属潜在的生物可利用性和环境迁移性(常燕等, 2015).Al、Ti、Fe、Cr和Ba在水相中的累积溶解度较小, 且Al、Fe和Ti在酸相中的溶解度增量只有水相的30%左右, 南海气溶胶中Al、Fe和Ti溶解度受环境pH影响并不大, 环境迁移性不明显.Cu、Zn溶解度受pH影响较大.Mn、Ni和Co水相累积溶解度较大, Co可达到55%, 但其酸相溶解度增量较少, 只占水相的30%左右, 其受外界pH影响不太大.Pb的酸溶出量是水溶出量的1.8倍, 水相溶解度为30%, “水+酸”溶解度可达84%, 增加近2倍, 外界pH变化对Pb溶解度控制明显, 当外界条件变化时, 特别是有酸雨时, 其环境迁移性大大增强.
分别以微循环淋溶法和连续淋溶法得到气溶胶金属溶解态浓度和累积溶解度, 在Milli-Q水相同淋溶时间条件下, 两者无显著性差异(t-检验, p>0.05).气溶胶中元素的溶解动力学取决于粒子的性质, 即元素的存在形态(Deguillaume et al., 2005).颗粒物表面易交换的金属能够被Milli-Q水快速溶出, 而与颗粒物晶体结合稳定的金属则比较难溶, 特别是Al、Fe、Ti地壳源元素, 多以硅酸盐结合形式存在, 稳定且不易释放, 质子需要不断碰撞才能将金属元素交换出来(Desboeufs et al., 1999), 即使是在pH=2的高浓度质子作用下, 也需要较长时间才能将其溶解释放(Stumm, 1997).微循环法溶液中存有相同的阳离子, 但由于外界溶液远没有达到饱和状态, 离子强度和同离子效应的影响可以忽略.颗粒物中人为源元素本身溶解度高(Degillaume et al., 2005), 在水相中溶出过程很快, 因而两种方法得到的溶解度没有显著性差异.
3.4 南海气溶胶干沉降通量大气微量元素沉降通量是生物地球化学模型的关键参数, 本研究采用下式计算微量元素的干沉降通量:
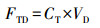
|
(4) |
式中, FTD为气溶胶中元素的总干沉降通量(μg · m-2 · d-1), CT为气溶胶中元素总浓度(μg · m-3), VD为气溶胶中元素的干沉降速度(m · s-1).
气溶胶中元素溶解态干沉降通量计算公式为:
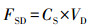
|
(5) |
式中, FSD为气溶胶中元素溶解态干沉降通量(μg · m-2 · d-1), CS为气溶胶中元素溶解态浓度(μg · m-3), VD为气溶胶中元素的干沉降速度(m · s-1).以上计算最大的误差(高达3倍)来源于VD的选择(Duce et al., 1991).干沉降速度对风速、相对湿度和颗粒物粒径的波动很敏感(Slinn et al., 1980).本文根据GESAMP推荐值、元素源分析结果及他人研究, 选取Al、Fe、Mn、Ti、Co地壳源为主的元素的沉降速率为1.0 cm · s-1 (Spokes et al., 2001), Cu、Ni、V、Zn、Pb等人为源为主的元素的沉降速率为0.1 cm · s-1(Chen et al., 2008).
南海夏季微量元素干沉降通量结果如图 6所示, 各元素FTD大小顺序为Al>Fe>Ti>Ba>Mn>Zn>Cr>V>Ni>Cu>Pb>Co, Al、Fe的FTD远高于其他元素, 平均值分别为(72.5±65.1)和(58.4±51.3) μg · m-2 · d-1.不同元素的总沉降通量中溶解态部分所占比例不同:Al、Fe和Ti的溶解态沉降通量在总沉降通量中仅占4%~6%, Cr和Ba为6%~7%, Cu、Zn和Pb为8%~17%, Mn、Co约为50%~60%.
 |
| 图 6 夏季南海气溶胶中微量元素溶解态、总干沉降通量盒状图(边界为25%和75%, 内黑线为中值;因样本容量不大, 所以没有展示离散范围) Fig. 6 Box diagrams of dissolvable and total dry deposition fluxes of trace elements in the aerosol samples of the South China Sea in summer |
南海气溶胶总干沉降通量显著小于其他边缘海(表 5), 一是因为夏季南海气溶胶来源主要为海源, 沙漠和人类活动源很少, 颗粒物浓度低;二是气溶胶运输时间和距离很长, 重力沉降作用明显, 粗颗粒很快沉降下来;三是采样期间有降雨过程, 降雨对颗粒物的清除作用很强.
| 表 5 不同海域微量元素总干沉降通量 Table 5 Total dry deposition fluxes of trace elements in different sea areas |
以Fe为例初步估算夏季南海东北部(18°~22°N, 112°~118°E)气溶胶沉降对初级生产的支持功能.对于颗粒物不同粒径, 其在海水中下沉速率为2~50 m · d-1(Fowler et al., 1986; Asper et al., 2003), 夏季南海气溶胶颗粒物多为细粒子, 选择沉降速率为10 m · d-1.夏季南海东北部混合层深度约为5~35 m(孙云鹏等, 2010), 则颗粒物离开混合层时间为0.5~3.5 d.南海溶解态Fe沉降通量为(2.4±1.4) μg · m-2 · d-1, 在离开上混合层的时间内, 气溶胶溶解态Fe输入量为1.2~8.4 μg · m-2, 即21.4~150.0 nmol · m-2.南海北部陆坡真光层溶解态Fe平均浓度为(0.50±0.17) nmol · L-1(Zhang et al., 2019), 则真光层单位面积溶解态Fe输入量为2500~16500 nmol · m-2, 大气气溶胶输入的溶解态Fe占南海北部溶解态Fe的0.13%~6.00%, 说明大气沉降对夏季南海初级生产有一定的支持功能.假设大气沉降的溶解态Fe能被海洋浮游植物完全利用, 以浮游植物细胞中Fe : C比值为8 μmol · mol-1计(Okin et al., 2011), 则观测期间大气沉降的溶解态Fe最多可支持南海(66±40) mg · m-2 · d-1的初级生产.夏季南海东北部的初级生产力约为262 mg · m-2 · d-1(于君等, 2016), 则大气沉降的溶解态Fe对南海东北部初级生产力的贡献约为25%, 比李鹏志等(2018)估计的青岛气溶胶对黄海初级生产力贡献值(10%)大得多.
4 结论(Conclusions)1) 夏季南海气溶胶微量元素溶解态浓度和总浓度在世界边缘海内处于较低水平, 但高于开阔大洋.气溶胶中Al、Fe、Mn、Ti、Co是以地壳源为主的元素, 人类活动添加量非常少;Ba、Cr有一定程度的富集, 为人类活动和地壳混合源;Cu、Zn、Pb、V、Ni以人类活动源为主.南海气溶胶颗粒在长距离运输过程中经历了充足的酸过程和云过程, 溶解度较大.
2) Milli-Q水和HCl溶液连续淋溶实验中, 气溶胶微量元素在Milli-Q水中25 min左右达到溶出平衡.Al、Fe、Ti的酸溶出量占总溶出量的23%, Mn、Ni和Co受pH影响不明显.环境pH对Ba、Pb溶解度控制明显, 酸溶出量可达水溶出量的近2倍.在相同时间内, 微循环淋溶和连续淋溶得到的微量元素溶解度没有显著性差异.
3) 南海夏季气溶胶总干沉降通量显著小于其他海域.大气沉降输入南海北部的可溶性Fe占南海北部溶解态Fe的0.13%~6.00%, 对海洋初级生产具有一定的支持功能.
Al-Taani A A, Rashdan M, Khashashneh S. 2015. Atmospheric dry deposition of mineral dust to the Gulf of Aqaba, Red Sea:Rate and trace elements[J]. Marine Pollution Bulletin, 92(1/2): 252-258. |
Asper V L, Smith W O. 2003. Abundance, distribution and sinking rates of aggregates in the Ross Sea, Antarctica[J]. Deep Sea Research Part I:Oceanographic Research Papers, 50(1): 131-150. DOI:10.1016/S0967-0637(02)00146-2 |
Baker A R, Croot P L. 2010. Atmospheric and marine controls on aerosol iron solubility in seawater[J]. Marine Chemistry, 120(1): 4-13. |
Bishop J K B. 2002. Robotic Observations of dust storm enhancement of carbon biomass in the north pacific[J]. Science, 298(5594): 817-821. DOI:10.1126/science.1074961 |
Bridgestock L, van de Flierdt T, Rehkamper M, et al. 2016. Return of naturally sourced Pb to Atlantic surface waters[J]. Nature Communications, 7: 12921. DOI:10.1038/ncomms12921 |
Browning T J, Bouman H A, Henderson G M, et al. 2014. Strong responses of Southern Ocean phytoplankton communities to volcanic ash[J]. Geophysical Research Letters, 41(8): 2851-2857. DOI:10.1002/2014GL059364 |
Buck C S, Landing W M, Resing J. 2013. Pacific Ocean aerosols:Deposition and solubility of iron, aluminum, and other trace elements[J]. Marine Chemistry, 157: 117-130. DOI:10.1016/j.marchem.2013.09.005 |
常燕, 冯冲, 瞿建国, 等. 2015. 上海大气总悬浮颗粒物中金属的可溶性特征[J]. 环境科学, 36(4): 1164-1172. |
Chen H, Chen L. 2008. Importance of anthropogenic inputs and continental-derived dust for the distribution and flux of water-soluble nitrogen and phosphorus species in aerosol within the atmosphere over the East China Sea[J]. Journal of Geophysical Research, 113(D11). DOI:10.1029/2007JD009491 |
Chester R, Murphy K J T, Lin F J, et al. 1993. Factors controlling the solubilities of trace metals from non-remote aerosols deposited to the sea surface by the 'dry' deposition mode[J]. Marine Chemistry, 42(2): 107-126. DOI:10.1016/0304-4203(93)90241-F |
Deguillaume L, Leriche M, Desboeufs K, et al. 2005. Transition metals in atmospheric liquid phases:Sources, reactivity, and sensitive parameters[J]. Chemical Reviews, 105(9): 3388-3431. DOI:10.1021/cr040649c |
Desboeufs K V, Losno R, Vimeux F, et al. 1999. The pH-dependent dissolution of wind-transported Saharan dust[J]. Journal of Geophysical Research:Atmospheres, 104(D17): 21287-21299. DOI:10.1029/1999JD900236 |
Deutsch F, Hoffmann P, Ortner H M. 1997. Analytical characterization of manganese in rainwater and snow samples[J]. Fresenius' Journal of Analytical Chemistry, 357(1): 105-111. DOI:10.1007/s002160050121 |
Doney S C, Fabry V J, Feely R A, et al. 2009. Ocean acidification:The other CO2 problem[J]. Annual Review of Marine Science, 1(1): 169-192. DOI:10.1146/annurev.marine.010908.163834 |
Duce R A, Liss P S, Merrill J T, et al. 1991. The atmospheric input of trace species to the world ocean[J]. Global Biogeochemical Cycles, 5(3): 193-259. DOI:10.1029/91GB01778 |
Eyckmans K, Zhang J, Hoog J D, et al. 2001. Leaching of nutrients and trace metals from aerosol samples:A comparison between a re-circulation and an ultrasound system[J]. International Journal of Environmental Analytical Chemistry, 80(3): 227-243. DOI:10.1080/03067310108044372 |
Fowler S W, Knauer G A. 1986. Role of large particles in the transport of elements and organic compounds through the oceanic water column[J]. Progress in Oceanography, 16(3): 147-194. |
Hsu S, Wong G T F, Gong G, et al. 2010. Sources, solubility, and dry deposition of aerosol trace elements over the East China Sea[J]. Marine Chemistry, 120(1/4): 116-127. |
Jickells T D, Baker A R, Chance R. 2016. Atmospheric transport of trace elements and nutrients to the oceans[J]. Philos Trans A Math Phys Eng Sci, 374(2081). DOI:10.1098/rsta.2015.0286 |
姬洪亮, 赵宏, 孔少飞, 等. 2011. 天津近岸海域大气颗粒物无机组分季节变化及源析[J]. 中国环境科学, 31(2): 177-185. |
Kadko D, Aguilar-Islas A, Bolt C, et al. 2019. The residence times of trace elements determined in the surface Arctic Ocean during the 2015 US Arctic GEOTRACES expedition[J]. Marine Chemistry, 208: 56-69. DOI:10.1016/j.marchem.2018.10.011 |
Koçak M, Nimmo M, Kubilay N, et al. 2004. Spatio-temporal aerosol trace metal concentrations and sources in the Levantine Basin of the Eastern Mediterranean[J]. Atmospheric Environment, 38(14): 2133-2144. DOI:10.1016/j.atmosenv.2004.01.020 |
Kumar P, Sokolik I N, Nenes A. 2011. Measurements of cloud condensation nuclei activity and droplet activation kinetics of fresh unprocessed regional dust samples and minerals[J]. Atmospheric Chemistry and Physics, 11(7): 3527-3541. DOI:10.5194/acp-11-3527-2011 |
李鹏志, 李茜, 石金辉, 等. 2018. 夏季青岛大气粗细粒子中微量元素的浓度、溶解度及干沉降通量[J]. 环境科学, 39(7): 3067-3074. |
Li T, Wang Y, Zhou J, et al. 2017. Evolution of trace elements in the planetary boundary layer in southern China:Effects of dust storms and aerosol-cloud interactions[J]. Journal of Geophysical Research Atmospheres, 122(6): 3492-3506. DOI:10.1002/2016JD025541 |
López-García P, Gelado-Caballero M D, Collado-Sánchez C, et al. 2017. Solubility of aerosol trace elements:Sources and deposition fluxes in the Canary Region[J]. Atmospheric Environment, 148: 167-174. DOI:10.1016/j.atmosenv.2016.10.035 |
Mahowald N M. 2005. Atmospheric global dust cycle and iron inputs to the ocean[J]. Global Biogeochemical Cycles, 19(4). DOI:10.1029/2004GB002402 |
Mahowald N M, Hamilton D S, Mackey K R M, et al. 2018. Aerosol trace metal leaching and impacts on marine microorganisms[J]. Nature Communications, 9(1): 2614-2629. DOI:10.1038/s41467-018-04970-7 |
Mahowald N M, Hamilton D S, Mackey K, et al. 2018. Aerosol trace metal leaching and impacts on marine microorganisms[J]. Nat Commun, 9(1): 2614. |
Meskhidze N, Chameides W L, Nenes A. 2005. Dust and pollution:A recipe for enhanced ocean fertilization?[J]. Journal of Geophysical Research-Atmospheres, 110(D3): 301. |
Okin G S, Baker A R, Tegen I, et al. 2011. Impacts of atmospheric nutrient deposition on marine productivity:Roles of nitrogen, phosphorus, and iron[J]. Global Biogeochemical Cycles, 25(2): 240-250. |
Qureshi S, Dutkiewicz V A, Khan A R, et al. 2006. Elemental composition of PM2.5 aerosols in Queens, New York:Solubility and temporal trends[J]. Atmospheric Environment, 40: 238-251. |
Saito M A, Goepfert T J, Ritt J T. 2008. Some thoughts on the concept of colimitation:Three definitions and the importance of bioavailability[J]. Limnology and Oceanography, 53(1): 276-290. DOI:10.4319/lo.2008.53.1.0276 |
Seinfeld J H, Pandis S N, Noone K.2006.Atmospheric Chemistry and Physics: From Air Pollution to Climate Change(2nd ed)[M].New York: Wiley, John & Sons.1203
|
Shelley R U, Roca-Martí M, Castrillejo M, et al. 2017. Quantification of trace element atmospheric deposition fluxes to the Atlantic Ocean (>40°N; GEOVIDE, GEOTRACES GA01) during spring 2014[J]. Deep Sea Research Part I:Oceanographic Research Papers, 119: 34-49. DOI:10.1016/j.dsr.2016.11.010 |
Shi G, Chen Z, Teng J, et al. 2012. Fluxes, variability and sources of cadmium, lead, arsenic and mercury in dry atmospheric depositions in urban, suburban and rural areas[J]. Environmental Research, 113: 28-32. DOI:10.1016/j.envres.2012.01.001 |
石金辉, 张云, 高会旺, 等. 2011. 东海大气气溶胶的化学特征及来源[J]. 环境科学学报, 31(8): 1750-1756. |
Shi J, Zhang J, Gao H, et al. 2013. Concentration, solubility and deposition flux of atmospheric particulate nutrients over the Yellow Sea[J]. Deep Sea Research Part II:Topical Studies in Oceanography, 97: 43-50. |
Sholkovitz E R, Sedwick P N, Church T M. 2009. Influence of anthropogenic combustion emissions on the deposition of soluble aerosol iron to the ocean:Empirical estimates for island sites in the North Atlantic[J]. Geochimica et Cosmochimica Acta, 73(14): 3981-4003. DOI:10.1016/j.gca.2009.04.029 |
Slinn S A, Slinn W G N. 1980. Predictions for particle deposition on natural waters[J]. Atmospheric Environment, 14(9): 1013-1016. DOI:10.1016/0004-6981(80)90032-3 |
Spokes L, Jickells T, Jarvis K. 2001. Atmospheric inputs of trace metals to the northeast Atlantic Ocean:the importance of southeasterly flow[J]. Marine Chemistry, 76(4): 319-330. DOI:10.1016/S0304-4203(01)00071-8 |
Stumm W. 1997. Reactivity at the mineral-water interface:dissolution and inhibition[J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 120(1): 143-166. |
孙云鹏, 张永刚, 金国栋.2010.南海上混合层厚度季节性变化及分析[C]."气象海洋环境与船舶航行安全"学术研讨会.大连: 3
|
Taylor S R. 1964. Abundance of chemical elements in the continental crust:A new table[J]. Geochimica et Cosmochimica Acta, 28(8): 1273-1285. DOI:10.1016/0016-7037(64)90129-2 |
Theodosi C, Markaki Z, Tselepides A, et al. 2010. The significance of atmospheric inputs of soluble and particulate major and trace metals to the eastern Mediterranean seawater[J]. Marine Chemistry, 120(1/4): 154-163. |
Uematsu M, Hattori H, Nakamura T, et al. 2010. Atmospheric transport and deposition of anthropogenic substances from the Asia to the East China Sea[J]. Marine Chemistry, 120(1/4): 108-115. |
Wayne D M, Diaz T A, Fairhurst R J, et al. 2006. Direct major- and trace-element analyses of rock varnish by high resolution laser ablation inductively-coupled plasma mass spectrometry (LA-ICPMS)[J]. Applied Geochemistry, 21(8): 1410-1431. DOI:10.1016/j.apgeochem.2006.04.005 |
吴兑. 1995. 南海北部大气气溶胶水溶性成分谱分布特征[J]. 大气科学, (5): 615-622. DOI:10.3878/j.issn.1006-9895.1995.05.11 |
吴天.2010.中国东海近海大气气溶胶的陆源特征、污染混合机理及干沉降量的估算[D].上海: 复旦大学.76 http://d.wanfangdata.com.cn/thesis/Y2225519
|
Wu Y, Zhang J, Liu S, et al. 2018. Aerosol concentrations and atmospheric dry deposition fluxes of nutrients over Daya Bay, South China Sea[J]. Marine Pollution Bulletin, 128: 106-114. DOI:10.1016/j.marpolbul.2018.01.019 |
于君, 邱永松. 2016. 黑潮入侵对南海东北部初级生产力的影响[J]. 南方水产科学, 12(4): 17-27. DOI:10.3969/j.issn.2095-0780.2016.04.003 |
张凯, 高会旺, 张仁健, 等. 2005. 我国沙尘的来源、移动路径及对东部海域的影响[J]. 地球科学进展, (6): 627-636. DOI:10.3321/j.issn:1001-8166.2005.06.005 |
Zhang R, Zhu X, Yang C, et al. 2019. Distribution of dissolved iron in the Pearl River(Zhujiang) Estuary and the northern continental slope of the South China Sea[J]. Deep Sea Research Part II:Topical Studies in Oceanography, 167: 14-24. DOI:10.1016/j.dsr2.2018.12.006 |
Zhang T, Shi J, Yao X, et al.2011.Sources, Solubility, and Deposition Fluxes of Trace Elements in Atmospheric Aerosols over the Yellow Sea[C].第七届亚洲气溶胶会议.西安: 940-944
|
Zhao R, Han B, Lu B, et al. 2015. Element composition and source apportionment of atmospheric aerosols over the China Sea[J]. Atmospheric Pollution Research, 6(2): 191-201. DOI:10.5094/APR.2015.023 |