 2021, Vol. 41
2021, Vol. 41



环境中芳香族化合物具有毒性大、可生化性差等特点, 是难降解工业废水中的主要污染物之一(Ryssel et al., 2015).臭氧工艺作为一种有效的水污染控制技术, 在饮用水消毒中的应用历史悠久, 近年来, 为去除污水中生物难降解的污染物, 臭氧化作为预处理工艺或深度处理工艺在难降解工业废水处理中的应用越来越广泛.然而臭氧化不能完全矿化有机物, 新生成的中间产物的生物毒性和积累潜力往往要高于母体化合物(Li et al., 2008; Gómez-Ramos et al., 2011; Kuang et al., 2013; Najjar et al., 2013).此外, 这些高毒性副产物可能比母体污染物更难去除, 并在环境中成为新的污染物(Hoigné et al., 1983; Yao et al., 1991; Shang et al., 2001; Turhan et al., 2007).因此, 有必要开展对臭氧化过程中生物毒性演变规律的研究, 为臭氧化工艺更加安全、高效的应用提供理论指导和技术支持.
以往对水处理中污染物去除的研究多集中在污染物的去除效率方面(陈行行等, 2017; 韩月等, 2019; Yang et al., 2020), 对生物毒性的变化关注较少, 尤其是对臭氧化中生物毒性的演变及高毒性中间产物的性质鲜有文献报道.受污染水体中污染物种类数以千万计, 传统的仅针对某些目标污染物浓度的测定, 对水质安全评价具有片面性.生物毒性测试法弥补了传统方法的不足, 可有效检测水体中所有共存污染物的综合生物效应, 能直观评价水质的安全性.发光菌在毒性测试中具有灵敏度高、操作简单等优点, 已广泛应用于反应样品的综合毒性和急性毒性的检测(马梅等, 1998; Jennings et al., 2001; Huang et al., 2011).
基于此, 本文以苯酚、邻甲酚、对甲酚、间甲酚、苯胺和对氯苯胺这6种芳香族化合物为研究对象, 从生态毒理学的角度评价臭氧化的效果, 即在评价污染物自身去除和TOC变化的同时, 研究其急性生物毒性在臭氧化过程中的演变, 揭示高毒性中间产物的性质及生成机制, 以期为臭氧化工艺的安全运行提供支撑, 从而提高水资源的安全性, 降低水质风险.
2 材料与方法(Materials and methods) 2.1 仪器与试剂主要仪器:高效液相色谱仪(HPLC, LC-20A, Shimadzu, 日本);臭氧发生器(CF-G-3-10G, 青岛国林, 中国);臭氧浓度检测仪(LT-200B, 青岛朗科, 中国);酶标分析仪(Infinite-200, Tecan, 瑞士);紫外分光光度计(Lambda-25, Vberlingen, 德国);TOC分析仪(Multi N/C 3100, Elementar, 德国);立式压力蒸汽灭菌锅(LDZX-75KBS, 上海申安,中国);恒温磁力搅拌器(C-MAG HS7, IKA, 德国).
6种芳香族化合物标准品:苯酚(phenol, BP)、邻甲酚(o-Cresol, o-C)、对甲酚(p-Cresol, p-C)、间甲酚(m-Cresol, m-C)、苯胺(aniline, AN)和对氯苯胺(p-Chloroaniline, p-CAN)均购自德国Aladdin Chemistry公司.高效液相色谱级叔丁醇(tert-Butanol, t-BuOH)购自中国Meryer Chemical公司;无水亚硫酸钠购自中国南京宁试化学试剂有限公司;碘化钾购自德国Aladdin Chemistry公司.
用于生物测试的KH2PO4、Na2HPO4 · 12H2O、MgSO4、NaHCO3、CaCl2、KCl等均为分析纯, 购自中国国药沪试化学试剂有限公司.
2.2 臭氧化实验在2.5 L的玻璃反应器中进行序批式臭氧化实验, 将反应器置于磁力搅拌器上, 通过磁子进行搅拌.纯氧通过臭氧发生器产生臭氧气体, 由臭氧发生器后连接的臭氧浓度检测仪对臭氧浓度进行检测, 待臭氧浓度指数在设定浓度下稳定后开始向反应器中进气, 以0.5 L · min-1的流速通过反应器底部的多孔曝气器均匀分散到溶液中, 尾气中的臭氧由尾端连接的KI溶液吸收.按一定时间间隔取水样, 取出后立即用氮气吹脱溶解在溶液中未反应的臭氧, 对水样进行母体污染物浓度、TOC和急性毒性测试, 研究初始污染物浓度、臭氧剂量、pH值和反应时间4个工艺条件对臭氧化后急性毒性的影响.初始污染物浓度为25~75 mg · L-1(p-CAN为34.2~102.6 mg · L-1), 臭氧剂量为14~42 mg · L-1. 在不同pH(3、5、7、10)下进行臭氧化反应, 不同pH的反应物溶液用10 mmol · L-1磷酸缓冲液配制, 再使用1 mol · L-1 H2SO4和1 mol · L-1 NaOH调节至相应pH值.向溶液中加入55 mmol · L-1叔丁醇, 以抑制羟基自由基与底物的反应.向1.2 mL臭氧化水样中加入37.3 mg · L-1亚硫酸钠, 用以去除氧化性物质, 考察其对急性毒性的贡献.水样于4 ℃保存, 48 h内分析完毕.
2.3 分析项目及检测方法 2.3.1 常规指标测定采用总有机碳分析仪测定水样TOC.采用配备紫外检测器(SPD-M20A)的高效液相色谱分析仪(HPLC)测定溶液中苯酚的浓度, 配备的色谱柱为改性C18柱(Inertsil ODS-SP, 250 mm×4.6 mm×5 μm).进样量为10 μL, 柱温为30 ℃, 流动相A为甲醇, 流动相B为超纯水, A与B的体积比为6.5 ∶ 3.5.流速为1 mL · min-1, 测试时间为10 min, 苯酚检测波长为271 nm, 保留时间为4.8 min.其他5种芳香族化合物的浓度采用紫外分光光度法测定, 采用双光束Lambda-25分光光度计在700~200 nm范围内扫描溶液紫外吸收光谱, 利用特征波长进行定量, 甲酚、苯胺和对氯苯胺的特征波长分别为271、280和290 nm.
2.3.2 发光菌急性毒性实验本实验受试菌种为青海弧菌Q67(Vibrio qinghaiensis sp.-Q67).实验方法参考已有研究(Ma et al., 1999), 具体方法如下:取在-80 ℃冰箱中保存的Q67菌种(1 mL菌液)接入装有液体培养基的锥形瓶, 于22 ℃、180 r · min-1条件下振荡培养16~18 h;然后取100 μL菌液于96孔板测定发光强度(RLU), 2000 r · min-1下离心10 min, 吸取菌体, 用模拟湖水将菌体制成菌悬液, 调整菌悬液的密度, 使其发光强度在20×104~60×104 RLU · mL-1之间待用.每次测试前, 将测试水样调整至pH为6.5~7.5, 并用模拟湖水稀释至一系列浓度.将180 μL水样及20 μL调整后的菌液加入96孔板中, 模拟湖水设置为空白对照, 水样的每个浓度设置3个平行.振荡15 min后, 用酶标仪测定各孔的发光强度.
采用稀释倍数的倒数表示样品的相对浓度, 根据式(1)计算发光抑制率IR.

|
(1) |
式中, RLIbc为空白对照组的发光强度;RLIs为稀释水样的发光强度.
使用Origin(Originlab, 2019b)拟合水样的剂量-效应曲线, 计算最大半抑制浓度, 具体如式(2)所示.
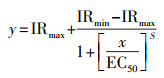
|
(2) |
式中, x为原水样在稀释水样中的百分比;y为呈现S形形状的发光抑制率;IRmax和IRmin分别为发光抑制率的最大值和最小值;S为斜率参数;EC50为最大半抑制浓度.
为了全面分析水样的毒性, 本文引入美国联邦环境保护局(USEPA)提出的毒性当量, 用毒性单位(TU)表示急性毒性(Shang et al., 2002; Han et al., 2019), 计算方法见式(3).
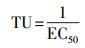
|
(3) |
母体污染物是臭氧化过程中高毒性中间产物生成的底物, 因此, 初始污染物浓度对臭氧化过程中急性毒性的演变至关重要(Wu et al., 2019).在pH为7、臭氧剂量为28 mg · L-1时, 研究初始污染物浓度对臭氧化6种芳香族化合物的降解率、矿化率与急性毒性变化的影响, 实验结果分别如图 1和2所示.
 |
| 图 1 不同初始污染物浓度下臭氧化苯酚(a)、邻甲酚(b)、对甲酚(c)、间甲酚(d)、苯胺(e)和对氯苯胺(f)中污染物浓度和TOC随反应时间的变化 Fig. 1 Changes in the concentrations of aromatics and TOC with time during the ozonation of BP(a), o-C(b), p-C(c), m-C(d), AN(e) and p-CAN(f) under the influence of initial aromatics concentrations |
 |
| 图 2 在不同初始污染物浓度下臭氧化苯酚(a)、邻甲酚(b)、对甲酚(c)、间甲酚(d)、苯胺(e)和对氯苯胺(f)中TU和TU(Na2SO3)随反应时间的变化 Fig. 2 Changes in TU and TU(Na2SO3) with time during the ozonation of BP(a), o-C(b), p-C(c), m-C(d), AN(e) and p-CAN(f) under the influence of initial aromatics concentrations |
由图 1可知, 随着初始污染物浓度增加, 臭氧化芳香族化合物的降解率均下降, 其中影响最大的为苯胺, 最终降解率从93.3%下降到72.9%;矿化率也均下降, 其中影响最大的为苯酚, 最终矿化率从55.6%下降到34.9%.
如图 2所示, 尽管母体污染物与TOC的浓度在臭氧化过程中持续下降, 但急性毒性随着反应时间的延长而明显增加, 直至达到最大值后下降.已有研究发现, 臭氧化氧四环素(Li et al., 2008)、甲氧苄啶(Kuang et al., 2013)、西替利嗪(Borowska et al., 2016)、苯扎贝特(Sui et al., 2017)、磺胺甲恶唑(Gómez-Ramos et al., 2011)和左氧氟沙星(Najjar et al., 2013)等化合物时生物毒性也出现了先升高后下降的变化规律.当初始污染物浓度增大, 臭氧化6种芳香族化合物过程中毒性单位最大值(TUmax)也随之增大, 这是因为污染物是生成高毒性中间产物的活性底物, 较高的初始污染物浓度意味着更多的底物参与了高毒性中间体的生成, 导致急性毒性增大, 此外, 也导致了毒性单位最大值出现时间的延后.如表 1所示, 毒性单位最大值(TUmax)与初始污染物浓度的对数值(lnC)呈线性关系, 可决系数R2为0.705~0.974.当4种酚类(苯酚、邻甲酚、对甲酚和间甲酚)的初始浓度增加至3倍时, 臭氧化后的TUmax增加了3倍及以上.其中影响最大的是邻甲酚, 当[o-C]=25 mg · L-1时, TUmax=8.1, 而当[o-C]=75 mg · L-1时, TUmax=51.4, 增加了5.3倍.导致生物毒性增加量高于初始污染物浓度增加量的原因可能是, 生成的高毒性中间产物浓度位于其剂量-效应曲线的线性变化范围, 或者生成的高毒性中间产物为多种, 它们存在协同的联合毒性作用(Liu et al., 2016).而两种胺类(苯胺和对氯苯胺)的初始浓度增加至3倍时, TUmax没有随母体污染物浓度等比例增加.这是因为随着初始苯胺和对氯苯胺浓度的增加, 苯环上的氨基通过释放更多的超氧阴离子自由基, 促进臭氧分解产生羟基自由基(Sarasa et al., 2002), 更多的羟基自由基可能导致有毒中间产物被迅速转化为低毒或无毒产物, 因此TUmax增加不明显.
| 表 1 初始污染物浓度的对数与TUmax的线性关系 Table 1 Linear relationship between logarithm of initial aromatics concentrations and TUmax |
如图 2所示, 臭氧化邻甲酚时急性毒性增长率最大, 当初始浓度为50 mg · L-1时, 前2 min增长率为11.4 TU · min-1, 而对甲酚增长率仅为1.5 TU · min-1. 由表 1可知, 当3种甲酚同分异构体初始浓度相同时, 臭氧化邻甲酚的TUmax最大, 对甲酚的TUmax最小, 约低2~3倍.由于甲基位置的不同, 导致了不同程度的共轭效应, 造成了甲酚苯环电子云密度的多样性(Yu et al., 2019).3种同分异构体中对甲酚结合度最大, 环上电子云密度最高, 最容易受到臭氧或羟基自由基的攻击.因此, 理论上臭氧化对甲酚过程中的急性毒性增长率最大, 然而实际上对甲酚的急性毒性增长率最小, TUmax最小, 推测可能是因为在臭氧化对甲酚过程中, 臭氧分子易于攻击羟基邻位(Valsania et al., 2012), 生成急性毒性较小的中间产物, 并且生成的有毒中间产物可能更容易被羟基自由基清除.邻甲酚苯环上电子云密度最低, 最难被臭氧和羟基自由基氧化.理论上臭氧化邻甲酚中急性毒性增长率最小, 但实际上邻甲酚的急性毒性增长率大于对甲酚, TUmax最大, 推测可能是臭氧分子攻击羟基对位, 生成的中间产物急性毒性大且较难被羟基自由基去除.间甲酚苯环上的电子云密度介于对甲酚和邻甲酚之间, 且间甲酚的对位和邻位同时被臭氧分子攻击, 因此, 臭氧化后的急性毒性增长率和TUmax也介于对甲酚和邻甲酚之间.
由表 1可知, 当苯胺和对氯苯胺初始物质的量浓度相同时, 臭氧化对氯苯胺的TUmax大于苯胺.推测可能是因为对氯苯胺对位上的氯取代基发生脱氯, 形成氯离子, 与臭氧分子反应生成自由氯(Shang et al., 2006), 导致急性毒性增加.
3.1.2 臭氧剂量对急性毒性演变的影响臭氧剂量对高毒性中间产物的形成有双重影响(Han et al., 2019):一方面, 臭氧是生成高毒性中间体的氧化剂;另一方面, 臭氧分子及其产物羟基自由基能够与高毒性中间产物继续反应以去除它们.在pH为7、初始污染物浓度不变时, 考察臭氧剂量从14 mg · L-1增加到42 mg · L-1对臭氧化芳香族化合物降解率、矿化率与急性毒性变化的影响, 实验结果分别如图 3和4所示.
 |
| 图 3 不同臭氧投加量下臭氧化苯酚(a)、邻甲酚(b)、对甲酚(c)、间甲酚(d)、苯胺(e)和对氯苯胺(f) 中污染物浓度和TOC随反应时间的变化 Fig. 3 Changes in the concentrations of aromatics and TOC with time during the ozonation of BP(a), o-C(b), p-C(c), m-C(d), AN(e) and p-CAN(f) under the influence of applied ozone dosages |
 |
| 图 4 不同臭氧剂量下臭氧化苯酚(a)、邻甲酚(b)、对甲酚(c)、间甲酚(d)、苯胺(e)和对氯苯胺(f) 中TU和TU(Na2SO3)随反应时间的变化 Fig. 4 Changes in TU and TU(Na2SO3) with time during the ozonation of BP(a), o-C(b), p-C(c), m-C(d), AN(e) and p-CAN(f) under the influence of applied ozone dosages |
由图 3可知, 臭氧剂量的增加提高了芳香族化合物的降解率, 其中影响最大的为间甲酚, 最终降解率从59.8%增加到92.2%;矿化率也均增加, 其中影响最大的为苯酚, 最终矿化率从38.3%增加到57.2%.
如图 4所示, 相比降解率和矿化率的变化, 生物毒性的变化表现出复杂性.当臭氧剂量为14、28和42 mg · L-1时, 臭氧化苯酚、间甲酚和对氯苯胺的TUmax呈下降趋势, 如图 4a、4d、4f所示, 其中影响最大的为间甲酚, 在[O3]=14 mg · L-1时, TUmax=31.6, 而当[O3]=42 mg · L-1时, TUmax=19.3, 下降了39%.较高的臭氧剂量会导致更多的有毒中间产物被去除, 从而TUmax变小.但如图 4b和4e所示, 随着臭氧剂量的增加, 邻甲酚和苯胺的TUmax呈升高趋势, 其中影响最大的为邻甲酚, 在[O3]=14 mg · L-1时, TUmax=25.3, 而当臭氧剂量增加到3倍时, TUmax=34.0, 增加了34.4%.这可能是由于增加的臭氧剂量主要与初始污染物反应, 导致高毒性中间产物生成的增加.如图 4c所示, 臭氧剂量的增加对臭氧化对甲酚的TUmax影响较小.臭氧与母体污染物反应的速率大于与高毒性中间产物的反应速率, 才会造成高毒性中间产物的累积.臭氧化过程中可能存在“临界臭氧剂量”(Beltran et al., 1990), 如果臭氧剂量低于“临界臭氧剂量”, 臭氧主要与母体污染物反应, 生成高毒性中间产物, 随着投加的臭氧剂量增大, 产生的高毒性中间体增多, 从而TUmax增大.如果臭氧剂量高于“临界臭氧剂量”, 此时臭氧主要与高毒性中间产物反应, 造成高毒性中间产物的去除, 随着投加的臭氧剂量增大, 更多的高毒性中间产物被去除, 造成TUmax下降.而这两方面的影响都会导致急性毒性峰值出现的时间提前(图 4).
由图 4和表 2可知, 臭氧化3种甲酚同分异构体时, 臭氧剂量的增加对它们的急性毒性的影响也不相同.邻甲酚是3种甲酚中最难被氧化的同分异构体, 因此它的“临界臭氧剂量”可能最大, 间甲酚次之, 对甲酚最小.对于相同浓度的甲酚, 臭氧剂量从14 mg · L-1增加到42 mg · L-1, 处理邻甲酚时TUmax呈升高趋势, 处理间甲酚时呈下降的趋势, 处理对甲酚时影响不大.
| 表 2 不同臭氧剂量下臭氧化6种芳香族化合物的TUmax Table 2 TUmax during the ozonation of 6 aromatics under the different ozone dosages |
由于氯取代基的增加, 在pH=7时, 对氯苯胺与臭氧的表观反应速率大于苯胺与臭氧的表观反应速率(Tekle-Röttering et al., 2016), 如图 3e和3f所示, 造成相同物质的量浓度下, “临界臭氧剂量”较小.因此, 对于相同物质的量浓度的苯胺和对氯苯胺, 臭氧剂量从14 mg · L-1增加到42 mg · L-1, 处理苯胺时TUmax呈升高趋势, 而处理对氯苯胺时TUmax呈下降趋势.
3.1.3 pH对急性毒性演变的影响pH在臭氧化过程中起着重要的作用, 因为pH不仅会影响水中臭氧的存在形式, 也会影响污染物的电离程度(Huang et al., 2020).本文研究了4个不同pH值(3、5、7和10)对臭氧化芳族化合物急性毒性演变的影响, 结果分别如图 5和6所示.
 |
| 图 5 在不同pH下臭氧化苯酚(a)、邻甲酚(b)、对甲酚(c)、间甲酚(d)、苯胺(e)和对氯苯胺(f) 中污染物浓度和TOC随反应时间的变化 Fig. 5 Changes in the concentrations of aromatics and TOC with time during the ozonation of BP(a), o-C(b), p-C(c), m-C(d), AN(e) and p-CAN(f) under the influence of solution pH |
 |
| 图 6 在不同pH下臭氧化苯酚(a)、邻甲酚(b)、对甲酚(c)、间甲酚(d)、苯胺(e)和对氯苯胺(f) 中TU和TU(Na2SO3)随反应时间的变化 Fig. 6 Changes in TU and TU(Na2SO3) with time during the ozonation of BP(a), o-C(b), p-C(c), m-C(d), AN(e) and p-CAN(f) under the influence of solution pH |
由图 5可知, 随着pH增大, 臭氧化芳香族化合物的降解率呈升高趋势, 矿化率也均提高, 其中, 影响最大的为苯酚, 最终矿化率从34.0%增加到67.9%.
由图 6可知, 随着pH增加, 臭氧化不同母体污染物的生物毒性变化规律不同.如图 6a~6d所示, 在臭氧化4种酚类化合物时, 随着反应溶液pH从3上升到7, 毒性单位最大值出现下降的趋势.这可能是因为随着pH的增大, 臭氧分子分解产生更多的羟基自由基, 它们会与高毒性中间产物反应, 导致毒性峰值变小.4种酚类化合物的pKa均大于10, 因此在pH=3~7时, 主要以非解离的分子形式存在.当pH增大到10时, pKa较低的苯酚(pKa=10.0)和间甲酚(pKa=10.1)电离程度显著增加, 与臭氧分子反应速率迅速增大, 导致了高毒性中间产物的积累, 从而TUmax又出现升高的现象.如图 6e和6f所示, 在臭氧化两种胺类化合物时, pH对TUmax的影响较小, pH=3~10时TUmax的相对标准偏差分别仅为18%和9.3%.这可能是因为在酸性条件下, 分子臭氧的作用是主导反应, 但两种胺类的氨基通过释放超氧阴离子自由基增强臭氧分解为羟基自由基.而在碱性条件下, 增加的氢氧根离子会促进羟基自由基的产生.另外, 当pH=3~10时, 苯胺和对氯苯胺均主要以质子化形式(pKa分别为4.6和4.0)存在, pH的增加对它们电离程度影响较小.以上两方面原因导致反应pH从3上升到10时, 臭氧化苯胺和对氯苯胺的TUmax变化较小.此外, 酸性条件下, 毒性达到TUmax的时间大于中性和碱性条件, 原因可能是随着pH的增大, 臭氧分子分解产生更多的羟基自由基, 提高了高毒性中间产物的降解速率, 缩短了达到TUmax的时间.
| 表 3 不同pH下臭氧化6种芳香族化合物的TUmax Table 3 TUmax during the ozonation of 6 aromatics under the different solution pH |
臭氧和有机化合物反应主要基于臭氧分子的直接反应和间接的羟基自由基反应(Hoigné et al., 1976; Nöthe et al., 2009; 李默等, 2018).为了揭示两种主要氧化剂对臭氧化芳香族化合物中急性生物毒性演变的影响, 在反应体系中加入羟基自由基清除剂叔丁醇.叔丁醇与臭氧分子几乎不发生反应(k(t-BuOH+O3)=0.003 L · mol-1 · s-1), 但与羟基自由基的反应速率非常快(k(t-BuOH+ · OH)=6×108 L · mol-1 · s-1), 因此, 常作为羟基自由基清除剂(刘朋朋等, 2010; 李学艳等, 2013; 王兵等, 2016). 研究发现, 在臭氧化叔丁醇的过程中, 生物毒性没有明显变化.在初始污染物浓度为50 mg · L-1 (p-CAN为68.4 mg · L-1)、pH为7、臭氧剂量为28 mg · L-1的条件下, 加入55 mmol · L-1叔丁醇时, 臭氧化6种芳香族化合物的急性毒性变化如图 7所示.由图 7可知, 臭氧化所有芳香族化合物的TUmax均显著增加, 增加最大的为苯酚, TUmax从9.6升高到93.6, 增加了8.8倍.这说明高毒性中间体主要是由芳香族化合物与臭氧分子反应生成, 而不是羟基自由基.另外, 说明在未添加叔丁醇的体系中, 羟基自由基能够与高毒性中间产物反应, 造成TUmax下降, 这与本研究前面的分析一致.
 |
| 图 7 在加入或不加入55 mmol · L-1叔丁醇的条件下臭氧化苯酚(a)、邻甲酚(b)、对甲酚(c)、间甲酚(d)、苯胺(e)和对氯苯胺(f) 中TU和TU(Na2SO3)随反应时间的变化 Fig. 7 Changes in TU and TU(Na2SO3) with or without 55 mmol · L-1 t-BuOH during the ozonation of BP(a), o-C(b), p-C(c), m-C(d), AN(e) and p-CAN(f) |
为了进一步揭示臭氧化芳香族化合物中高毒性中间产物的性质, 在反应后的溶液中加入还原剂Na2SO3, 研究生物毒性的变化规律.如图 2、图 4、图 6和图 7所示, 还原剂Na2SO3能够高效地去除累积的高毒性中间产物, 造成生物毒性的显著下降.对于4种酚类化合物, 37.3 mg · L-1的Na2SO3最高能够去除134.9 TU的生物毒性, 对TUmax的去除率为96.1%.但对于两种胺类, 37.3 mg · L-1的Na2SO3最高能够去除36.8 TU的生物毒性, 对TUmax的去除率为83.3%.由此推测, 臭氧化酚类化合物时, 高毒性中间产物均具有氧化性, 是导致急性毒性增加的主要原因, 而臭氧化胺类化合物时, 少部分高毒性中间产物不具有氧化性.氧化性物质在空气中可能被还原, 这与已有研究(Petala et al., 2006)中, 臭氧化水样在储存48 h后发生了部分急性毒性降低的现象一致.芳香族化合物在臭氧化中会产生中间产物醌类, 醌类不稳定, 能够产生半醌自由基和活性氧自由基等, 是一种重要的有机氧化剂.醌类的氧化性会导致生物毒性作用, 被认为是造成苯酚等污染物臭氧化过程中生物毒性升高的重要原因(Shang et al., 2001; Xu et al., 2018).醌类化合物在芳香族化合物臭氧化中对生物毒性的贡献还需要进一步的研究.
高毒性中间产物这一性质的揭示有利于后续开发针对性的还原工艺, 以达到对臭氧化芳香族化合物过程中生物毒性的控制.
4 结论(Conclusions)1) 初始污染物浓度、臭氧剂量、pH和反应时间等工艺条件会影响苯酚、邻甲酚、对甲酚、间甲酚、苯胺和对氯苯胺这6种芳香族化合物臭氧化时生物毒性的变化.初始污染物浓度增大, 臭氧化芳香族化合物的生物毒性最大值增大.随着臭氧剂量增大, 臭氧化苯酚、间甲酚和对氯苯胺的生物毒性最大值下降, 臭氧化邻甲酚和苯胺的生物毒性最大值升高, 臭氧化对甲酚的生物毒性最大值变化较小.随着pH从3升高到7, 臭氧化4种酚类化合物时生物毒性最大值呈下降趋势;pH对臭氧化苯胺和对氯苯胺生物毒性最大值的影响较小.
2) 臭氧分子是臭氧化芳香族化合物时高毒性中间产物生成的主要氧化剂.
3) 氧化性高毒性中间产物是臭氧化过程中急性毒性升高的主要贡献物, 这一性质的揭示有利于指导开发针对性的生物毒性控制方法, 以达到臭氧工艺的安全运行.
Beltran F J, Encinar J M, Garcia-Araya J F. 1990. Ozonation of o-Cresol in aqueous solution[J]. Water Research, 24(11): 1309-1316. DOI:10.1016/0043-1354(90)90146-W |
Borowska E, Bourgin M, Hollender J, et al. 2016. Oxidation of cetirizine, fexofenadine and hydrochlorothiazide during ozonation: kinetics and formation of transformation products[J]. Water Research, 94: 350-362. DOI:10.1016/j.watres.2016.02.020 |
陈行行, 白智勇, 李群, 等. 2017. 臭氧氧化降解水中三氯乙烯的效能研究[J]. 环境科学学报, 37(12): 4586-4592. |
Gómez-Ramos M M, Mezcua M, Agüera A, et al. 2011. Chemical and toxicological evolution of the antibiotic sulfamethoxazole under ozone treatment in water solution[J]. Journal of Hazardous Materials, 192(1): 18-25. |
韩月, 任逸, 翟姝, 等. 2019. 污水生化出水有机物对氯贝酸臭氧降解的影响研究[J]. 环境科学学报, 39(11): 3779-3785. |
Han Q, Dong W Y, Wang H J, et al. 2019. Degradation of tetrabromobisphenol a by ozonation: Performance, products, mechanism and toxicity[J]. Chemosphere, 235: 701-712. DOI:10.1016/j.chemosphere.2019.06.204 |
Hoigné J, Bader H. 1976. The role of hydroxyl radical reactions in ozonation processes in aqueous solutions[J]. Water Research, 10(5): 377-386. DOI:10.1016/0043-1354(76)90055-5 |
Hoigné J, Bader H. 1983. Rate constants of reactions of ozone with organic and inorganic compounds in water-Ⅰ: non-dissociating organic compounds[J]. Water Research, 17(2): 173-183. DOI:10.1016/0043-1354(83)90098-2 |
Huang W Y, Liu F, Liu S S, et al. 2011. Predicting mixture toxicity of seven phenolic compounds with similar and dissimilar action mechanisms to Vibrio qinghaiensis sp.nov.Q67[J]. Ecotoxicology & Environmental Safety, 74(6): 1600-1606. |
Huang Y, Su L H, Zhang S Y, et al. 2020. Opposite pH-dependent roles of hydroxyl radicals in ozonation and UV photolysis of genistein[J]. Science of the Total Environment, 709. DOI:10.1016/j.scitotenv.2019.136243 |
Jennings V L K, Rayner-Brandes M H, Bird D J. 2001. Assessing chemical toxicity with the bioluminescent photobacterium (vibrio fischeri): A comparison of three commercial systems[J]. Water Research, 35(14): 3448-3456. DOI:10.1016/S0043-1354(01)00067-7 |
Kuang J M, Huang J, Wang B, et al. 2013. Ozonation of trimethoprim in aqueous solution: identification of reaction products and their toxicity[J]. Water Research, 47(8): 2863-2872. DOI:10.1016/j.watres.2013.02.048 |
李默, 汪震哲, 陈志强, 等. 2018. AAO工艺联合臭氧削减污水中微量有机污染物及遗传毒性[J]. 环境科学, 39(10): 4584-4592. |
李学艳, 王永喜, 黄勇, 等. 2013. 臭氧氧化降解水中四丁基锡效能及动力学研究[J]. 环境科学学报, 33(5): 1272-1277. |
刘朋朋, 张华, 石锐, 等. 2010. 蜂窝陶瓷催化臭氧化苯乙酮的研究[J]. 环境科学学报, 30(10): 2043-2048. |
Li K X, Yediler A, Yang M, et al. 2008. Ozonation of oxytetracycline and toxicological assessment of its oxidation by-products[J]. Chemosphere, 72(3): 473-478. DOI:10.1016/j.chemosphere.2008.02.008 |
Liu J Q, Qu R J, Yan L Q, et al. 2016. Evaluation of single and joint toxicity of perfluorooctane sulfonate and zinc to Limnodrilus hoffmeisteri: acute toxicity, bioaccumulation and oxidative stress[J]. Journal of Hazardous Materials, 301: 342-349. DOI:10.1016/j.jhazmat.2015.09.010 |
马梅, 童中华, 王子健, 等. 1998. 新型淡水发光菌(Vibrio qinghaiensis sp.-Q67)应用于环境样品毒性测试的初步研究[J]. 环境科学学报, 18(1): 86-91. |
Ma M, Tong Z H, Wang Z J, et al. 1999. Acute toxicity bioassay using the freshwater luminescent bacterium vibrio-qinghaiensis sp.Nov.-Q67[J]. Bulletin of Environmental Contamination & Toxicology, 62(3): 247-253. |
Najjar N H E, Touffet A, Deborde M, et al. 2013. Levofloxacin oxidation by ozone and hydroxyl radicals: kinetic study, transformation products and toxicity[J]. Chemosphere, 93(4): 604-611. DOI:10.1016/j.chemosphere.2013.05.086 |
Nöthe T, Fahlenkamp H, Sonntag C V. 2009. Ozonation of wastewater: Rate of ozone consumption and hydroxyl radical yield[J]. Environmental Science & Technology, 43(15): 5990-5995. |
Petala M, Samaras P, Zouboulis A, et al. 2006. Ecotoxicological properties of wastewater treated using tertiary methods[J]. Environmental Toxicology, 21(4): 417-424. DOI:10.1002/tox.20188 |
Ryssel S T, Arvin E, LützhØft H H, et al. 2015. Degradation of specific aromatic compounds migrating from PEX pipes into drinking water[J]. Water Research, 81: 269-278. DOI:10.1016/j.watres.2015.05.054 |
Sarasa J, Cortés S, Ormad P, et al. 2002. Study of the aromatic by-products formed from ozonation of anilines in aqueous solution[J]. Water Research, 36(12): 3035-3044. DOI:10.1016/S0043-1354(02)00003-9 |
Shang N C, Yu Y H. 2001. The biotoxicity and color formation results from ozonation of wastewaters containing phenol and aniline[J]. Journal of Environmental Science and Health. Part A, Toxic/Hazardous Substances & Environmental Engineering, 36(3): 383-393. |
Shang N C, Yu Y H, Ma H W. 2002. Variation of toxicity during the ozonation of monochlorophenolic solutions[J]. Journal of Environmental Science and Health. Part A, Toxic/Hazardous Substances & Environmental Engineering, 37(2): 261-271. |
Shang N C, Yu Y H, Ma H W, et al. 2006. Toxicity measurements in aqueous solution during ozonation of mono-Chlorophenols[J]. Journal of Environmental Management, 78(3): 216-222. DOI:10.1016/j.jenvman.2005.03.015 |
Sui Q, Gebhardt W, Schr der H F, et al. 2017. Identification of new oxidation products of bezafibrate for better understanding of its toxicity evolution and oxidation mechanisms during ozonation[J]. Environmental Science & Technology, 51(4): 2262-2270. |
Tekle-Röttering A, Sonntag C V, Reisz E, et al. 2016. Ozonation of anilines: kinetics, stoichiometry, product identification and elucidation of pathways[J]. Water Research, 98: 147-159. DOI:10.1016/j.watres.2016.04.001 |
Turhan K, Uzman S. 2007. The Degradation Products of Aniline in the Solutions with Ozone and Kinetic Investigations[J]. Annali Di Chimica, 97(10): 1129-1138. DOI:10.1002/adic.200790096 |
Valsania M C, Fasano F, Richardson S D, et al. 2012. Investigation of the degradation of cresols in the treatments with ozone[J]. Water Research, 46(8): 2795-2804. DOI:10.1016/j.watres.2012.02.040 |
王兵, 周鋆, 任宏洋, 等. 2016. MgO催化臭氧氧化降解苯酚机理研究[J]. 环境科学学报, 36(11): 4009-4016. |
Wu Q Y, Zhou Y T, Li W X, et al. 2019. Underestimated risk from ozonation of wastewater containing bromide: both organic byproducts and bromate contributed to the toxicity increase[J]. Water Research, 162: 43-52. DOI:10.1016/j.watres.2019.06.054 |
Xu J L, Yu Y, Ding K, et al. 2018. Heterogeneous catalytic ozonation of hydroquinone using sewage sludge-derived carbonaceous catalysts[J]. Water Science & Technology, 77(5): 1410-1417. |
Yang J X, Luo C, Li T T, et al. 2020. Superfast degradation of refractory organic contaminants by ozone activated with thiosulfate: efficiency and mechanisms[J]. Water Research, 176. DOI:10.1016/j.watres.2020.115751 |
Yao C C D, Haag W R. 1991. Rate constants for direct reactions of ozone with several drinking water contaminants[J]. Water Research, 25(7): 761-773. DOI:10.1016/0043-1354(91)90155-J |
Yu L, Han P W, Jin H B, et al. 2019. Catalytic ozonation of three isomeric cresols in the presence of NaCl with nano-mesoporous β-molecular sieves[J]. Process Safety & Environmental Protection, 129: 63-73. |