 2021, Vol. 41
2021, Vol. 41



2. 工业聚集区污染控制与生态修复教育部重点实验室, 广州 510006
2. The Key Laboratory of Pollution Control and Ecosystem Restoration in Industry Clusters, Ministry of Education, Guangzhou 510006
近年来, 水体镉污染事件时有发生, 导致流域镉超标, 影响河流生态系统, 威胁当地及下游居民的安全.应对水体镉污染事故的措施之一是向水体中投入大量的聚合硫酸铁(PFS)形成絮体, 通过混凝使Cd2+沉淀到底泥中.异化铁还原菌广泛分布于沉积物等天然厌氧环境中.课题组前期的研究表明异化铁还原菌可以还原解构负载Cd的PFS絮体, 导致其结合的Cd释放(Li et al., 2016), 故镉污染事故应急处理中, 沉入底泥的镉可能会释放并再分配.
微生物异化铁还原过程受多种因素影响, 如电子穿梭体(腐殖质、黄素等溶解性有机质)可加速微生物和矿物间的电子传递, 有利于铁还原(Frankel et al., 2003; Piepenbrock et al., 2011; Chen et al., 2015);环境中共存的金属离子如锰、铬、砷可被部分异化铁还原菌还原(Refait et al., 2017), 可能会对异化Fe(Ⅲ)还原过程产生竞争抑制作用;环境中的阴离子会占据铁矿物的表面位点抑制微生物对铁矿物的异化铁还原过程, 另一方面, 当环境中阴离子浓度较高时, 阴离子会参与次生矿物的形成, HCO3-会促进菱铁矿形成, PO43-有助于产生蓝铁矿(Fredrickson et al., 1998; Wang et al., 2019).
胞外聚合物(Extracellular polymeric substances, EPSs)的分泌是自然环境中微生物的普遍属性, 在原核生物和真核生物中均有发生.EPSs是有机大分子的混合物, 主要由蛋白质、多糖、脂质、核酸等物质构成(Sheng et al., 2010;More et al., 2014).EPSs作用广泛, 可与金属离子发生离子交换、吸附络合、氧化还原反应(王亮等, 2010).对于矿物, 一方面, EPSs的有机组分可以提供质子或者与矿物表面形成配合物促进矿物分解, 另一方面, EPSs含有羧基、羟基等大量活性官能团, 可提供丰富的离子吸附和矿物成核中心, 固定和富集环境中的金属离子, 促进矿物形成(Mann et al., 1991;Konhauser, 1998).
但目前关于EPSs对微生物异化铁还原过程的影响尚不清楚, 因此本课题组开展了EPSs对典型异化铁还原菌Shewanella oneidensis MR-1还原含镉聚合硫酸铁(Cd-PFS)的影响研究.本研究发现有助于深入理解胞外聚合物对微生物异化铁还原这一生物地球化学过程的影响, 在实际应用中, 也为Cd-PFS絮体在真实沉积物环境中可能受到的影响及Cd释放风险提供了理论指导.
2 材料与方法(Materials and methods) 2.1 絮体的制备方法称取10 g聚合硫酸铁(PFS)至烧杯中, 溶解后定容配成10%的聚合硫酸铁溶液.取出20 mL聚合硫酸铁溶液加入至500 mL浓度为7.48 mg·L-1的Cd(NO3)2溶液中, 进行快速搅拌混凝, 用5 mol·L-1的NaOH溶液将混合液的pH值调至7.2.搅拌结束后, 将絮体静置, 絮体沉降至200 mL左右, 倒掉上清液, 保存絮体备用.用混合酸(HNO3、HF、HClO4)将絮体消解后, 用原子吸收分光光度法测得Cd-PFS絮体中的Cd含量为13.04 mg·L-1.
2.2 菌体的培养与富集Shewanella oneidensis MR-1购买于美国模式菌种培养库(700550).Luria-Bertani(LB)培养基购买于阿拉丁, Defined Medium(DM)培养基(Smeaton et al., 2009)的成分为:1.34 mmol·L-1 KCl, 28 mmol·L-1 NH4Cl, 0.68 mmol·L-1 CaCl2·2H2O, 50 mmol·L-1 NaClO4, 20 mmol·L-1 1, 4-哌嗪二乙磺酸(1, 4-piperazine diethanesulfonic acid, PIPES)和24 mmol·L-1乳酸钠.用1 mol·L-1 NaOH将LB培养基和DM培养基pH值调至7.2.在超净台内, 从斜管培养基中先将S. oneidensis MR-1接种至LB液体培养基中, 并在150 r·min-1、30 ℃条件下好氧培养15 h;再把LB液体培养基中的菌体接种至平板培养基中, 保存在4 ℃冰箱, 使用时将S. oneidensis MR-1从平板培养基接种至LB液体培养基中, 按上述方法再次培养活化.离心收集菌体, 用DM培养基清洗两次, 对菌体称重后重悬于DM培养基中, 配制成一定浓度的细菌悬浮液(Li et al., 2016).
2.3 EPSs的提取采用甲醛/NaOH法提取Shewanella. oneidensis MR-1的EPSs (罗曦等, 2005;Liu et al., 2013).Shewanella. oneidensis MR-1在LB培养基中培养15 h之后, 500 mL菌液中加入3 mL 36.5%甲醛, 放入4 ℃冰箱反应, 甲醛可以使细胞稳定, 减少细胞溶解, 1 h后菌液中加入200 mL 1 mol·L-1 NaOH, 放入4 ℃冰箱反应3 h, 之后在10000 r·min-1转速下高速离心10 min.采用NaOH提取了紧密结合态EPSs后, 通过高速离心, 上清液中同时包括了紧密结合态EPSs和松散附着态EPSs.弃掉细菌后, 用0.22 μm滤膜过滤EPSs溶液以去除溶液中残留的细菌.随后EPSs溶液放入1000截留分子量透析袋中进行透析以去除溶液中的低分子量杂质, 每24 h更换一次超纯水, 透析72 h后, 将EPSs溶液冷冻干燥48 h即得到EPSs粉末, 将EPSs粉末保存备用.
2.4 序批实验实验在摇床中振荡进行.将EPSs粉末溶解在DM培养基中, 配置成不同浓度EPSs溶液.准确量取2 mL Cd-PFS絮体、20 mL细菌悬液分装至体积为100 mL的顶空瓶中, 再添加0.5 mL不同浓度EPSs溶液, 顶空瓶中EPSs终浓度分别为0.03 g·L-1和0.3 g·L-1(EPSs是环境中天然有机质的重要组成, Cai等研究指出自然环境中天然有机质浓度范围为几百μg·L-1至几百mg·L-1 (Cai et al., 2018);本研究将S. oneidensis MR-1菌液培养15 h后, 每1 g细菌可提取0.254 g EPSs.根据文献、EPSs提取产率及设置的细菌初始浓度(1 g·L-1), 本研究设置了实验组的EPSs浓度分别为0.03 g·L-1和0.3 g·L-1);Cd-PFS+S. oneidensis MR-1对照组中加入2 mL Cd-PFS絮体、20.5 mL细菌悬液;Cd-PFS+killed S. oneidensis MR-1对照组中加入2 mL Cd-PFS絮体和20.5 mL己高温高压灭活的S. oneidensis MR-1细菌悬液;Cd-PFS+0.3 g·L-1 EPSs对照组中加入2 mL Cd-PFS絮体和20.5 mL EPSs溶液, 顶空瓶中EPSs终浓度为0.3 g·L-1, 所有有菌体系中菌的最终浓度为1 g·L-1.配置好的Cd-PFS絮体的Cd2+浓度为13.04 mg·L-1, 各组中Cd2+终浓度为1.16 mg·L-1.同时向顶空瓶中不断充入高纯N2, 以去除顶空瓶顶部的氧气, 确保体系处于厌氧状态, 然后用含有丁基硅胶的铝盖快速密封顶空瓶, 最后放置于30 ℃, 转速为150 r·min-1的摇床中避光反应.分别于0、24、48、72、120、168、288、480、960 h取样, 每个时间点设置3个平行样, 并对反应样品进行化学分析和固相表征.
2.5 生物矿化模拟实验利用EPSs作为矿化模板进行矿化模拟实验, 分别将0.012 g、0.12 g EPSs粉末溶解在100 mL PIPES缓冲溶液中(pH=7.2), 在微生物异化铁还原过程中Fe3+是逐渐溶解释放的, 为了模拟该过程, 用蠕动泵将20 mL FeCl3溶液逐滴加入EPSs溶液中, 滴速为0.0035 mL·min-1.最终体系中Fe3+浓度为300 mg·L-1, EPSs浓度分别为0.1、1 g·L-1;Fe3+ +PIPES缓冲对照组则将20 mL FeCl3溶液逐滴加入100 mL PIPES缓冲溶液中.滴加完成后, 静置20 d进行矿化反应, 在特定时间点取样测试溶液中的Fe3+浓度和体系pH, 20 d后取样进行固相表征.
2.6 EPSs吸附实验分别将0.1 mL 0、4.5、45 mg·L-1 Fe3+溶液中加入10 mL 0.1 g·L-1 EPSs溶液中, 然后立即搅拌均匀, 用0.5 mol·L-1 HCl将pH调至1.5以避免Fe3+发生沉淀, 室温下静置2 h以达到吸附平衡, 之后取溶液进行三维荧光光谱测试(3D-EEM).
2.7 分析方法序批实验中采用邻菲罗啉分光光度法在波长为510 nm条件下测定用6 mol·L-1盐酸溶液提取的可萃取态Fe(Ⅱ)浓度(Borch et al., 2007), 具体步骤如下: 取1 mL样品溶液, 加入3 mL 6 mol·L-1盐酸, 在30 ℃, 转速150 r·min-1的条件下避光振荡提取1 h, 经孔径为0.22 μm的水相滤膜过滤后进行测定(曾桃, 2017).测定溶液中的Fe3+浓度时, 将顶空瓶中溶液轻轻摇匀后用灭菌注射器取出2 mL样品, 经0.22 μm孔径水相滤头过滤后, 取滤液测定溶液中Fe2+及总Fe的浓度, 总Fe浓度减去Fe2+浓度即得到溶液中Fe3+的浓度.溶液中的Cd2+浓度测定:取5 mL样品溶液, 经0.22 μm孔径水相滤头过滤, 用5%硝酸酸化稀释后, 采用原子吸收分光光度计检测.生物矿化模拟实验中, 将样品离心并过0.22 μm孔径水相滤头, 用5%硝酸酸化稀释后, 采用ICP-OES测定上清液中Fe3+浓度, 同时测定其pH.固相表征前, 将样品溶液离心, 离心后的固体沉淀用蒸馏水清洗两次以去除残留的培养基, 将离心后的沉淀真空冷冻干燥24 h后研磨成粉末进行固相表征.采用荷兰PANalytical公司Empyrean型X射线衍射仪测定矿物的组成和结晶度.采用Zeiss公司Merlin型场发射扫描电镜获取矿物形貌图像, 利用能谱分析(EDS)测定了矿物表面主要元素的分布.采用德国Bruker公司EQUINOX55型傅里叶变换红外光谱仪测定固相样品化学基团或化学键变化.采用美国Thermo Fisher公司Escalb 250Xi型X-射线光电子能谱仪获取固相表面的构成元素、原子价态和原子之间的成键情况.对EPSs进行了成分分析, 以葡萄糖为标准, 采用蒽酮法测定EPSs中多糖的含量(Gaudy et al., 1962).以牛血清白蛋白为标准, 采用Bradford法测定EPSs中蛋白含量(Bradford, 1976).采用紫外可见分光光度计测定EPSs中DNA的含量(Boonaert et al., 2001).
采用日本Hitachi公司F-7000型分子荧光光谱仪测定了EPSs与Fe3+反应前后的EEM荧光光谱.扫描波长范围设置为: 激发波长(Ex)250~400 nm, 发射波长(Em)300~550 nm, 步长为10 nm.在分析之前, 将超纯水的光谱记录为空白, 并从样品的EEM光谱中扣除.
3 结果与讨论(Results and discussion) 3.1 EPSs促进微生物还原Fe(Ⅲ)图 1a显示了6 mol·L-1盐酸可提取态Fe(Ⅱ)浓度随时间的变化.研究表明, 6 mol·L-1 HCl可以提取体系中的所有Fe(Ⅱ)(Swanner et al., 2017), 可以代表还原产生的Fe2+含量.细菌灭活组由于S. oneidensis MR-1失去活性, 因此溶液中始终检测不到Fe(Ⅱ)的产生.120 h时, 0.03和0.3 g·L-1 EPSs实验组中的Fe(Ⅱ)浓度分别达到15.3和33.63 mg·L-1, 而Cd-PFS+S. oneidensis MR-1对照组(未外加EPSs)Fe(Ⅱ)浓度仅为4.2 mg·L-1.168 h后, 0.03和0.3 g·L-1 EPSs实验组Fe(Ⅱ)浓度分别是Cd-PFS+S. oneidensis MR-1对照组的1.65倍和2.72倍.288 h后, 3组Fe(Ⅱ)浓度分别达到85.77、92.9和102.53 mg·L-1, 然后保持相对稳定.本研究还探究了单独EPSs对絮体的还原作用, 结果显示72 h, Cd-PFS+0.3 g·L-1 EPSs对照组中Fe(Ⅱ)浓度为6.4 mg·L-1, 与Cd-PFS+S. oneidensis MR-1+0.3 g·L-1 EPSs实验组中的Fe(Ⅱ)浓度(6.43 mg·L-1)接近, 高于其他组中的Fe(Ⅱ)浓度, 120 h, 3个外加EPSs组中的Fe(Ⅱ)浓度均高于未外加EPSs的Cd-PFS+S. oneidensis MR-1对照组, 而168 h后, Cd-PFS+0.3 g·L-1 EPSs对照组中的Fe(Ⅱ)浓度逐渐趋于稳定, 不再增加, 含有S. oneidensis MR-1活菌的反应体系Fe(Ⅱ)浓度持续升高, 远超Cd-PFS+0.3 g·L-1 EPSs对照组, 实验结果表明EPSs加快了Fe(Ⅲ)的还原, 反应前期(120 h前), 主要是外加的EPSs促进铁还原, 反应后期(168 h后), 细菌逐渐分泌了较多的EPSs, 分泌的EPSs也开始在铁还原过程中发挥促进作用.EPSs能够还原絮体中的Fe (Ⅲ), 可能是由于EPSs含有还原酶及糖类(王亮等, 2010), Sanghi等对杂色云芝胞外Ag纳米颗粒合成进行了研究, 指出胞外还原酶和糖类促成了Ag+向Ag转化, 其中—SH基团起着关键作用(Sanghi et al., 2009).本研究测定了溶液中的Fe3+随时间的变化趋势, 如图 1b所示, 各组溶液中Fe3+的浓度始终处于较低的水平(小于15.6 mg·L-1), 这是由于S. oneidensis MR-1、EPSs将溶解到溶液中的Fe3+不断地还原为Fe2+以及中性条件下Fe3+发生了沉淀作用.
 |
| 图 1 盐酸可萃取态Fe(Ⅱ)(a);溶液中的Fe3+浓度(b);溶液中的Cd2+浓度(c)随时间变化趋势 Fig. 1 The change in the HCl extractable Fe (Ⅱ) concentration (a), aqueous Fe3+ (b) and aqueous Cd2+ concentration(c) as a function of time |
本研究测定了溶液中的Cd2+随时间的变化趋势, 如图 1c所示, 反应初期(24 h前), 除细菌灭活组外, 其余4组溶液中的Cd2+逐渐增加, 这是由于微生物或EPSs还原Cd-PFS絮体中的Fe(Ⅲ), 导致絮体解构, 使得絮体吸附的Cd2+释放至溶液中.由于EPSs促进了铁还原, 故外加EPSs实验组中Cd2+的释放速率更快, 释放量更多, 24 h Cd-PFS+S. oneidensis MR-1对照组、Cd-PFS+0.3 g·L-1EPSs对照组、0.03 g·L-1 EPSs实验组、0.3 g·L-1 EPSs实验组溶液中的Cd2+浓度分别达到0.33、0.37、0.35和0.40 mg·L-1.之后, 溶液中的Cd2+浓度逐渐下降, 这是由于次生矿物在形成过程中, 重新吸附、络合了溶液中的Cd2+.从图中可以看出, 反应后期, EPSs实验组溶液中的Cd2+始终低于对照组, 这可能是由于EPSs促进了次生矿物的形成进而促进了Cd2+的重新吸附.细菌灭活组溶液中Cd2+浓度由0 h时的0.30 mg·L-1持续下降至960 h时的0.10 mg·L-1, 下降的原因可能是由于失活的细菌菌体表面吸附了溶液中的Cd2+.实验结果表明EPSs可能通过影响铁还原速率及次生矿物形成过程而影响Cd2+的再分配.
3.2 EPSs促进次生矿物形成对反应前的Cd-PFS絮体及10、20、40 d时各组的固样进行了XRD表征(图 2a~2c), XRD结果显示反应前的Cd-PFS絮体呈非晶态, 10 d后, Cd-PFS+0.3 g·L-1 EPSs对照组、Cd-PFS+S. oneidensis MR-1对照组和0.03 g·L-1 EPSs实验组固样仍呈无定形结构, 而0.3 g·L-1 EPSs实验组产生了纤铁矿、磁铁矿及蓝铁矿.20 d后, 0.3 g·L-1 EPSs实验组矿物衍射峰强度增强, 结晶度升高, 而其他3组矿物依然处于非晶态.40 d后, 0.03 g·L-1 EPSs实验组也出现了纤铁矿、磁铁矿及蓝铁矿, 而Cd-PFS+0.3 g·L-1 EPSs对照组和Cd-PFS+S. oneidensis MR-1对照组中仍未发现结晶矿物.EPSs实验组矿物的衍射图样符合JCPDS卡片纤铁矿(No.44-1415)、磁铁矿(No.19-0629)、蓝铁矿(No.03-0070)给出的衍射图样.
 |
| 图 2 矿物XRD图(a. 10 d, b. 20 d, c. 40 d)及SEM图 d. 反应前的Cd-PFS形貌, e. Cd-PFS+0.3 g·L-1 EPSs对照组, f. Cd-PFS+S. oneidensis MR-1对照组, g. 0.03 g·L-1 EPSs实验组, h. 0.3 g·L-1 EPSs实验组)(L=纤铁矿、M=磁铁矿、V=蓝铁矿 Fig. 2 XRD patterns of minerals at 10 d(a), 20 d (b), and 40 d(c) and SEM image of initial Cd-PFS(d); SEM images of minerals formed with 0.3 g·L-1 EPSs(e), S. oneidensis MR-1(f), S. oneidensis MR-1 +0.03 g·L-1 EPSs (g), and S. oneidensis MR-1+0.3 g·L-1 EPSs(h) at 40 d (L=Lepidocrocite, M=Magnetite, and V=vivianite) |
我们采用SEM-EDS检测了固样的表面形貌和元素组成.图 2d为反应前的Cd-PFS絮体的SEM图像, Cd-PFS絮体表面较为光滑平整, 呈无定形结构, 图 2e~2h显示了40 d后不同组的固样形貌, 两个EPSs实验组(图 2g~2h)中均出现了形貌良好的高结晶度矿物, 板状或片状矿物为纤铁矿, 粒状或块状且铁含量高于纤铁矿(见表 1元素组成)的矿物为磁铁矿(徐龙乾, 2018), 而Cd-PFS+0.3 g·L-1对照组中仍为与Cd-PFS絮体形貌、组成相似的无定形铁矿物(图 2e), Cd-PFS+S. oneidensis MR-1对照组矿物表面由光滑变粗糙(图 2f), 但仍呈无定形结构.SEM-EDS结果与XRD结果一致, EPSs促进了高结晶度次生矿物形成.
| 表 1 反应前Cd-PFS絮体及第40 d不同组固样各分析点Fe、O、S、P元素原子数百分比 Table 1 Elemental percentages of Fe, O, S, and P at each analysis spot of initial Cd-PFS and minerals at 40 d |
XRD结果表明, 添加EPSs促进了纤铁矿、磁铁矿的生成.纤铁矿及磁铁矿形成可能是由于EPSs提供了大量活性位点以固定溶液中的铁离子, 促进了铁矿物的形成(如式(1)~(2)所示).这个结论与前人研究结果一致, Bao等报道了Purpureocillium lilacinum strain Y3分泌的EPSs可作为结晶位点促进黄钾铁矾形成(Bao et al., 2018).
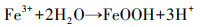
|
(1) |
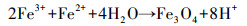
|
(2) |
为了证实EPSs可作为模板促进铁矿物的形成, 本研究利用EPSs作为矿化模板进行了矿化模拟实验, 分别将0.012、0.12 g EPSs粉末溶解在100 mL PIPES缓冲溶液中(pH=7.2), 将20 mL FeCl3溶液逐滴加入EPSs溶液中; Fe3+ +PIPES缓冲对照组则将20 mL FeCl3溶液逐滴加入100 mL PIPES缓冲溶液中.滴加完成后, 静置20 d进行矿化反应.
本研究检测了溶液中的Fe3+随时间的变化趋势, 如图 3a所示, EPSs实验组Fe3+浓度下降明显快于Fe3++PIPES缓冲对照组, 48 h后, 1 g·L-1 EPSs和0.1 g·L-1 EPSs组溶液中的Fe3+分别下降了64.2%与8.0%, 但是Fe3++PIPES缓冲对照组中的Fe3+没有下降.240 h后, 1 g·L-1 EPSs组、0.1 g·L-1 EPSs组、Fe3++PIPES缓冲对照组中的Fe3+分别下降至30.63、136.33及186.33 mg·L-1, 之后保持相对稳定.实验结果表明EPSs加快了铁沉淀速度, 提高了成矿效率.
 |
| 图 3 溶液中的Fe3+(a)及pH(b)随时间变化趋势 Fig. 3 The change in the aqueous Fe3+ concentration(a) and pH(b) as a function of time |
如图 3b所示, 随着反应进行, 3组的pH均逐渐降低.pH的降低是由于铁水解及矿化过程中H+的产生.溶液中的Fe3+水解会产生H+, 酸化环境, 铁离子沉淀为铁氢氧化物也会释放H+, 进一步降低pH.铁氢氧化物的形成过程如式(3)、式(4)所示:
首先, Fe3+发生水解:
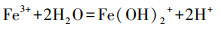
|
(3) |
然后, 水解后的Fe3+沉淀为铁氢氧化物(Chan et al., 2004):
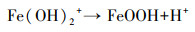
|
(4) |
本课题组的研究结果与前人研究一致.Warren等报道了胞外聚合物的存在促进了在过饱和程度较低的情况下形成铁氢氧化物(Warren et al., 1998).最初的吸附阶段过后, 表面位点达到饱和, 一旦达到临界过饱和, 就会立即发生沉淀.研究者认为在EPSs表面进行沉淀大大降低了界面自由能(ΔGi).
20 d时取固样进行了XRD检测, 如图 4a所示, 所有组中都产生了针铁矿和纤铁矿(由于矿化模拟实验未加入Fe2+溶液且未进行厌氧操作, 故矿物中没有出现Fe(Ⅲ)/Fe(Ⅱ)矿物磁铁矿), 矿物的衍射图样符合JCPDS卡片纤铁矿(No.44-1415)及针铁矿(No.29-0713)给出的衍射图样.从吸收峰强度增强以及衍射位点增加可以看出, EPSs的存在显著提高了矿物的结晶度, 其中, 0.1 g·L-1 EPSs实验组形成的矿物结晶度最高.1 g·L-1 EPSs实验组形成的矿物吸收峰相对于0.1 g·L-1 EPSs实验组矿物较弱, 结晶度较差, 可能是1 g·L-1 EPSs实验组中过多的活性官能团瞬间吸附了大量的铁离子, 在EPSs上形成了空间相互分离的Fe(Ⅲ)中心(González et al., 2014), 溶液中剩余的Fe3+较少, 不利于矿物的生长, 所以最终形成的矿物结晶度低于0.1 g·L-1 EPSs实验组矿物.
 |
| 图 4 20 d矿物XRD图谱(a), SEM图(b.EPSs, c.Fe3++PIPES缓冲对照组, d.Fe3+0.1 g·L-1 EPSs实验组, e.Fe3+1 g·L-1 EPSs实验组(L=纤铁矿;G=针铁矿)), EPSs及20 d矿物的红外光谱(f)及不同铁浓度组的三维荧光光谱(g.0 mg·L-1 Fe3+, h.4.5 mg·L-1 Fe3+, i.45 mg·L-1 Fe3+) Fig. 4 XRD patterns of minerals at 20 d(a); SEM images of unreacted EPSs(b), and minerals formed at 20 d with PIPES buffer control group (c), 0.1 g·L-1 EPSs(d) and 1 g·L-1 EPSs (e) (L=lepidocrocite and G=goethite); FTIR spectra of unreacted EPSs and minerals at 20 d(f); and 3D-EEM fluorescence spectra with different Fe3+ concentrations (g.0 mg·L-1 Fe3+, h.4.5 mg·L-1 Fe3+, i.45 mg·L-1 Fe3+) |
对固样进行了SEM检测, 如图 4b~4e所示, EPSs实验组, 尤其是0.1 g·L-1 EPSs实验组中形成了更多高结晶度且形貌良好的针铁矿和纤铁矿, SEM结果与XRD结果相符.
本研究对EPSs进行了成分分析, EPS中含有53.21%蛋白质, 20.36%多糖及10.29%DNA, EPSs组成见表 2.并对EPSs及矿物进行了FTIR检测.EPSs红外光谱波段归属如下:1657 cm-1处的波段为酰胺Ⅰ官能团(振动主要来自蛋白质C=O带的平面拉伸), 1589 cm-1附近为酰胺Ⅱ官能团(主要是蛋白质的N─H弯曲耦合C─N拉伸)(Jiang et al., 2004;Cao et al., 2011), 1404 cm-1处的波段归因于蛋白质、脂肪酸和氨基酸中的羧基的C─O对称拉伸, 1318、1252和983 cm-1处的波段是由于核酸或磷酸化蛋白质中的磷酸二酯键的不对称伸缩振动引起的(Adamou et al., 2016), 多糖(脂多糖/肽聚糖)的C─O、C─C、C─O─C和C─O─P伸缩产生的一系列复杂而有顺序的波段出现在1200~900 cm-1.
| 表 2 Shewanella oneidensis MR-1的EPSs组成 Table 2 Constituents of EPS extracted from Shewanella oneidensis MR-1 |
酰胺Ⅰ常用于表示蛋白质的二级结构变化(Haris et al., 1999).如图 4f所示, 20 d后酰胺Ⅰ从1657 cm-1处位移至1640 cm-1处, 且波段变宽.酰胺Ⅰ频率变化与蛋白质构象结构从随机无序结构(1660~1650 cm-1)变为α-螺旋结构(1650~1640 cm-1)有关, 表明由于蛋白质吸附了铁导致蛋白质构象发生了改变(Buijs et al., 1996).这个变化说明EPSs中的蛋白质参与到了EPSs诱导的成矿过程(Fang et al., 2012).发现反应后, 1404 cm-1处羧基的强度大大减弱, 这是由于Fe3+、Fe2+直接与EPSs蛋白质中的羧基结合造成的, 表明EPSs蛋白质中的羧基参与了对铁的吸附.反应前的EPSs在920 cm-1处具有尖锐的吸收峰, 920 cm-1处的波段归属于O─P─O的对称拉伸振动, 而反应20 d后, 形成的矿物在此处的吸收峰变小, 可能是Fe与EPSs中的磷酸基官能团发生了络合.反应后, 归属于vasPO2的波段发生了显著变化, 位于1318、1252和983 cm-1处归属于磷酸二酯键中的vasPO2的吸收峰强度显著减弱, 这是由于磷酸二酯键中的vasPO2与铁发生络合使得磷原子的电子密度减小且游离的P=O基缺失所导致的(Barja et al., 1999), 最重要的是, 在1022 cm-1处出现了新的吸收峰, 该吸收峰是P─O─Fe的伸缩振动引起的(Omoike et al., 2004), 这些波段的变化(强度改变、新增)表明来自EPSs核酸及磷酸化蛋白质中的磷酸基作为成核位点, 与溶液中的铁发生内层配位, 与铁原子形成P─O─Fe键, 促进了针铁矿及纤铁矿的形成.
另外, 本研究还进行了EPSs吸附Fe3+实验.达到吸附平衡后, 进行了三维荧光光谱测试, 图 4g~4i显示了不同铁浓度组的三维荧光光谱, 激发/发射波长(Ex/Em)为275~280/345~350 nm的峰为蛋白质中的芳香氨基酸色氨酸(Baker, 2001).如图 4g~4i所示, 反应后该峰的荧光强度随铁浓度增加而减弱, 这是因为蛋白质中的官能团与溶液中的Fe3+发生了吸附络合作用使得蛋白质发生荧光淬灭(Wingender et al., 1999).这个结果再次表明EPSs中的蛋白质在固定及聚集Fe3+的过程中发挥了重要作用, 有助于铁的沉淀, 在生物矿化过程可促进铁矿物形成.Wang等也报道了水环境中的类蛋白质分子大大增加了Fe3O4磁性纳米颗粒的聚集(Wang et al., 2018).
3.4 EPSs影响Cd迁移转化为进一步探索由于EPSs诱导的高结晶度矿物形成是否会对Cd2+的再吸附产生重要影响, 采用X射线光电子能谱检测方法对体系中的C、O、Cd 3种化学元素进行了分析.在XPS Peak 4.1软件中进行分峰拟合, 图 5a为0 h C1s元素图谱, 观察到C1s元素图谱中拟合出3个峰, 查文献可知, 284.8 eV为C─C/C─H;286.34 eV为C─O(醇羟基或醚基);288.23 eV为C─OOH(羧基)(Leng et al., 2015).图 5b为480 h C1s元素图谱, 从图中可以看出, 与0 h相比, 此时3个组中C─O峰面积比均下降, 这表明了醚基吸附了Cd2+, 形成了醚-镉络合物, 其中醚基中的氧将电子提供给Cd2+, 因此相邻两个碳原子的电子密度降低, 导致C─O峰面积比下降(Sheng et al., 2004).
 |
| 图 5 不同组固相样品XPS分析图谱(a.0 h C1s, b.480 h C1s, c.0 h O1s, d. 480 h O1s) Fig. 5 XPS spectra of solid-phase samples in different groups (a. 0 h C1s, b. 480 h C1s, c. 0 h O1s, d. 480 h O1s) |
图 5c为0 h O1s元素图谱, 此时O1s元素图谱中可拟合出3个峰, 其中, 532.1 eV为C─O;531.1 eV为C=O;529.7 eV为Fe─O.图 5d为480 h O1s元素图谱, 从图中可看出, 480 h时, C─O峰结合能从532.10 eV增强至533.0 eV, 这是由于镉离子和醚基中的氧络合, 使得氧原子的结合能增强(Huang et al., 2012).另外, 结合能为531.7 eV处出现了新峰, 该新峰归属于Fe─OH晶格(Baltrusaitis et al., 2007), 表明反应形成了铁氢氧化物, 与XRD结果相符.
图 6a为0 h Cd 3d元素图谱, 此时Cd 3d元素图谱中可拟合出4个峰, 其中, 405.4 eV和412.0 eV为Cd2+(Boparai et al., 2013), 指示初始时聚铁絮体吸附的Cd2+;另一对较小的峰位于404.8 eV和411.6 eV处, 指示Cd2+与醚基、醇羟基、羧基等官能团相结合形成的Cd─O(He et al., 2014).图 6b为480 h Cd 3d元素图谱, 从图中可以看出, EPSs实验组中, 405.4 eV和412.0 eV处的Cd2+峰面积比减小, 而404.8 eV和411.6 eV处的Cd─O峰面积比增大.尤其是在高浓度EPSs组中, 405.4 eV和412.0 eV处的Cd2+峰面积比显著减小, 而404.8 eV和411.6 eV处的Cd─O峰面积比显著增大, 这是由于EPSs促进了蓝铁矿、纤铁矿、磁铁矿等矿物的形成, 而这些矿物中大量的羟基为Cd2+的固定提供了氧原子(Ponthieu et al., 2006).Cd-PFS + S. oneidensis MR-1对照组中没有发生这种变化, 镉仍然主要以矿物表面吸附态形式Cd2+存在.这些结果表明, 反应后Cd-PFS+S. oneidensis MR-1对照组中, 被释放到溶液中的Cd2+由于静电作用吸附于不定形矿物表面, 为外层静电吸附, 而EPSs实验组中, 溶液中的Cd2+主要吸附于羟基吸附位点(≡Fe─OH), 为内层络合, 这种结合形式对Cd2+有更强的结合能力, 因此对Cd2+有更强的束缚性, 极大地减弱了Cd2+的可迁移性和可生化性.
 |
| 图 6 不同组固相样品XPS分析图谱(a.0 h Cd 3d, b. 480 h Cd 3d) Fig. 6 XPS spectra of solid-phase samples in different groups (a. 0 h Cd 3d, b. 480 h Cd 3d) |
结合上述实验结果, 提出了EPSs对次生矿物形成及Cd再分配的影响机制, 如图 7所示, EPSs中的磷酸基可作为成核位点, 固定和聚集溶液中的Fe3+、Fe2+, 促进纤铁矿、磁铁矿等铁矿物形成, 而这些高结晶度铁矿物含有丰富的羟基官能团, 有利于吸附固定溶液中的Cd2+.
 |
| 图 7 胞外聚合物对次生矿物形成及镉再分配的影响机制图 Fig. 7 Schematic diagram of the effects of EPSs on secondary mineral formation and cadmium redistribution |
胞外聚合物中磷酸化蛋白质和核酸中的磷酸基可作为成核位点, 与铁原子形成P—O—Fe键, 固定和聚集溶液中的Fe3+、Fe2+, 加速铁的沉淀, 促进纤铁矿、磁铁矿等铁矿物形成.胞外聚合物加快铁还原的同时会促使矿物负载的镉释放到溶液中, 但是同时由于胞外聚合物促进铁矿物形成, 铁矿物含有丰富的羟基官能团, 对Cd2+有很强的吸附能力, 最终有利于降低Cd2+的可迁移性及可生化性.
Adamou A, Manos G, Messios N, et al. 2016. Probing the whole ore chalcopyrite-bacteria interactions and jarosite biosynthesis by Raman and FTIR microspectroscopies[J]. Bioresource Technology, 214: 852-855
|
Baker A. 2001. Fluorescence Excitation-Emission Matrix Characterization of Some Sewage-Impacted Rivers[J]. Environmental Science & Technology, 35(5): 948-953. |
Baltrusaitis J, Cwiertny D M, Grassian V H. 2007. Adsorption of sulfur dioxide on hematite and goethite particle surfaces[J]. Physical Chemistry Chemical Physics, 9(41): 5542-5554. DOI:10.1039/b709167b |
Bao P, Xia M, Liu A, et al. 2018. Extracellular polymeric substances (EPS) secreted by Purpureocillium lilacinum strain Y3 promote biosynthesis of jarosite[J]. RSC Advances, 8(40): 22635-22642. DOI:10.1039/C8RA03060J |
Barja B C, Tejedor-Tejedor M I, Anderson M A. 1999. Complexation of Methylphosphonic Acid with the Surface of Goethite Particles in Aqueous Solution[J]. Langmuir, 15(7): 2316-2321. DOI:10.1021/la980540y |
Boonaert C J P, Dufrêne Y F, Derclaye S R, et al. 2001. Adhesion of Lactococcus lactis to model substrata: direct study of the interface[J]. Colloids and Surfaces B: Biointerfaces, 22: 171-182. DOI:10.1016/S0927-7765(01)00196-5 |
Boparai H K, Joseph M, O'Carroll D M J E S, et al. 2013. Cadmium (Cd2+) removal by nano zerovalent iron: surface analysis, effects of solution chemistry and surface complexation modeling[J]. Environmental Science and Pollution Research, 20(9): 6210-6221. DOI:10.1007/s11356-013-1651-8 |
Borch T, Fendorf S. 2007. Phosphate interactions with iron (hydr) oxides: Mineralization pathways and phosphorus retention upon bioreduction Environmental Science and Pollution Research[J]. Developments in Earth and Environmental Sciences, 7: 321-348. |
Bradford M M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding[J]. Analytical Biochemistry, 72: 248-254. DOI:10.1016/0003-2697(76)90527-3 |
Buijs J, Norde W, Lichtenbelt J W T. 1996. Changes in the secondary structure of adsorbed IgG and F(ab')2 Studied by FTIR Spectroscopy[J]. Langmuir, 12(6): 1605-1613. DOI:10.1021/la950665s |
Cai L, Hu L, Shi H, et al. 2018. Effects of inorganic ions and natural organic matter on the aggregation of nanoplastics[J]. Chemosphere, 197: 142-151. DOI:10.1016/j.chemosphere.2018.01.052 |
Cao B, Ahmed B, Kennedy D W, et al. 2011. Contribution of extracellular polymeric substances from shewanella sp. HRCR-1 Biofilms to U(Ⅵ) Immobilization[J]. Environmental Science & Technology, 45(13): 5483-5490. |
Chan C S, De Stasio G, Welch S A, et al. 2004. Microbial polysaccharides template assembly of nanocrystal fibers[J]. Science, 303: 5664, 1656. |
Chen C, Kukkadapu R, Sparks D L. 2015. Influence of coprecipitated organic matter on Fe2+(aq)-Catalyzed transformation of ferrihydrite: Implications for carbon dynamics[J]. Environmental Science & Technology, 49(18): 10927-10936. |
Fang L, Cao Y, Huang Q, et al. 2012. Reactions between bacterial exopolymers and goethite: Acombined macroscopic and spectroscopic investigation[J]. Water Research, 46(17): 5613-5620. DOI:10.1016/j.watres.2012.07.046 |
Frankel R B, Bazylinski D A. 2003. Biologically Induced Mineralization by Bacteria[J]. Reviews in Mineralogy and Geochemistry, 54(1): 95-114. DOI:10.2113/0540095 |
Fredrickson J K, Zachara J M, Kennedy D W, et al. 1998. Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium[J]. Geochimica et Cosmochimica Acta, 62: 3239-3257. DOI:10.1016/S0016-7037(98)00243-9 |
Gaudy E, Wolfe R S. 1962. Composition of an extracellular poly-saccharide produced by Sphaerotilus natans[J]. Applied Microbiology, 10: 200-205. DOI:10.1128/AM.10.3.200-205.1962 |
González A G, Pokrovsky O S, Jiménez-Villacorta F, et al. 2014. Iron adsorption onto soil and aquatic bacteria: XAS structural study[J]. Chemical Geology, 372: 32-45. DOI:10.1016/j.chemgeo.2014.02.013 |
Haris P I, Severcan F. 1999. FTIR spectroscopic characterization of protein structure in aqueous and non-aqueous media[J]. Journal of Molecular Catalysis B: Enzymatic, 7(1): 207-221. |
He J S, Chen J P. 2014. A comprehensive review on biosorption of heavy metals by algal biomass: Materials, performances, chemistry, and modeling simulation tools[J]. Bioresource Technology, 160: 67-78. DOI:10.1016/j.biortech.2014.01.068 |
Huang J, Ye M, Qu Y, et al. 2012. Pb (Ⅱ) removal from aqueous media by EDTA-modified mesoporous silica SBA-15[J]. Journal of Colloid and Interface Science, 385(1): 137-146. DOI:10.1016/j.jcis.2012.06.054 |
Jiang W, Saxena A, Song B, et al. 2004. Elucidation of functional groups on gram-positive and gram-negative bacterial surfaces using infrared spectroscopy[J]. Langmuir, 20(26): 11433-11442. DOI:10.1021/la049043+ |
Konhauser K O. 1998. Diversity of bacterial iron mineralization[J]. Earth-Science Reviews, 43(3): 91-121. |
Leng L, Yuan X, Zeng G, et al. 2015. Surface characterization of rice husk bio-char produced by liquefaction and application for cationic dye (Malachite green) adsorption[J]. Fuel, 155: 77-85. DOI:10.1016/j.fuel.2015.04.019 |
Li C, Yi X, Dang Z, et al. 2016. Fate of Fe and Cd upon microbial reduction of Cd-loaded polyferric flocs by Shewanella oneidensis MR-1[J]. Chemosphere, 144: 2065-2072. DOI:10.1016/j.chemosphere.2015.10.095 |
Liu X, Eusterhues K, Thieme J, et al. 2013. STXM and NanoSIMS investigations on EPS fractions before and after adsorption to goethite[J]. Environmental Science & Technology, 47(7): 3158-3166. |
罗曦, 雷中方, 张振亚, 等. 2005. 好氧/厌氧污泥胞外聚合物(EPS)的提取方法研究[J]. 环境科学学报, 25(12): 1624-1629. DOI:10.3321/j.issn:0253-2468.2005.12.010 |
Mann S. 1988. Molecular Recognition in Biomineralization[J]. Nature, 332(6160): 119-124. DOI:10.1038/332119a0 |
More T T, Yadav J S S, Yan S, et al. 2014. Extracellular polymeric substances of bacteria and their potential environmental applications[J]. Journal of Environmental Management, 144: 1-25. |
Omoike A, Chorover J, Kwon K D, et al. 2004. Adhesion of bacterial exopolymers to α-FeOOH: Inner-sphere complexation of phosphodiester groups[J]. Langmuir, 20(25): 11108-11114. DOI:10.1021/la048597+ |
Piepenbrock A, Dippon U, Porsch K, et al. 2011. Dependence of microbial magnetite formation on humic substance and ferrihydrite concentrations[J]. Geochimica et Cosmochimica Acta, 75(22): 6844-6858. DOI:10.1016/j.gca.2011.09.007 |
Ponthieu M, Juillot F, Hiemstra T, et al. 2006. Metal ion binding to iron oxides[J]. Geochimica et Cosmochimica Acta, 70(11): 2679-2698. DOI:10.1016/j.gca.2006.02.021 |
Refait P, Sabot R, Jeannin M. 2017. Role of Al(Ⅲ) and Cr(Ⅲ) on the formation and oxidation of the Fe(Ⅱ-Ⅲ) hydroxysulfate Green Rust[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 531: 203-212. |
Sanghi R, Verma P. 2009. Biomimetic synthesis and characterisation of protein capped silver nanoparticles[J]. Bioresource Technology, 100(1): 501-504. DOI:10.1016/j.biortech.2008.05.048 |
Sheng G P, Yu H Q, Li X Y. 2010. Extracellular polymeric substances (EPS) of microbial aggregates in biological wastewater treatment systems: A review[J]. Biotechnology Advances, 28(6): 882-894. DOI:10.1016/j.biotechadv.2010.08.001 |
Sheng P X, Ting Y P, Chen J P, et al. 2004. Sorption of lead, copper, cadmium, zinc, and nickel by marine algal biomass: characterization of biosorptive capacity and investigation of mechanisms[J]. Journal of Colloid and Interface Science, 275(1): 131-141. DOI:10.1016/j.jcis.2004.01.036 |
Smeaton C M, Fryer B J, Weisener C G. 2009. Intracellular precipitation of Pb by Shewanella putrefaciens CN32 during the reductive dissolution of Pb-jarosite[J]. Environmental Science & Technology, 43(21): 8086-8091. |
Swanner E D, Bayer T, Wu W, et al. 2017. Iron isotope fractionation during Fe(Ⅱ) oxidation mediated by the oxygen-producing marine cyanobacterium synechococcus PCC 7002[J]. Environmental Science & Technology, 51(9): 4897-4906. |
Wang H, Zhao X, Han X, et al. 2018. Colloidal stability of Fe3O4 magnetic nanoparticles differentially impacted by dissolved organic matter and cations in synthetic and naturally-occurred environmental waters[J]. Environmental Pollution, 241: 912-921. DOI:10.1016/j.envpol.2018.06.029 |
Wang Q, Wei Z, Yi X, et al. 2019. Biogenic iron mineralization of polyferric sulfate by dissimilatory iron reducing bacteria: Effects of medium composition and electric field stimulation[J]. Science of the Total Environment, 684: 466-475. DOI:10.1016/j.scitotenv.2019.05.322 |
Warren L A, Ferris F G. 1998. Continuum between sorption and precipitation of Fe(Ⅲ) on Microbial Surfaces[J]. Environmental Science & Technology, 32(15): 2331-2337. |
Wingender J, Neu T R, Flemming H C. 1999. What are Bacterial Extracellular Polymeric Substances?[M]. Berlin, Heidelberg: Springer Berlin Heidelberg.
|
王亮, 陈桂秋, 曾光明, 等. 2010. 真菌胞外聚合物及其与重金属作用机制研究进展[J]. 环境污染与防治, 32(6): 74-80. DOI:10.3969/j.issn.1001-3865.2010.06.017 |
徐龙乾. 2018. 电絮凝法处理高盐冶炼废水中重金属及机制研究[D]. 昆明: 昆明理工大学
|
曾桃. 2017. 微生物异化还原含Cd聚合硫酸铁絮体过程中影响因素的研究[D]. 广州: 华南理工大学
|