 2021, Vol. 41
2021, Vol. 41



2. 宁波市华研节能环保安全设计研究有限公司, 宁波 315800
2. Ningbo Huayan Energy Efficiency Environmental Protection Safety Design and Research Co., Ltd., Ningbo 315800
随着科技水平的提高, 人为排放挥发性有机物(Volatile Organic Compounds, VOCs)明显增多, 其主要来源与机动车排放的尾气、化石燃料的燃烧过程、石油精炼、石油产品的储存及分配、工业排放和溶剂的使用有关(Subrahmanyam et al., 2010).VOCs不仅具有“三致”作用, 还会导致光化学烟雾的发生, 从而引起了社会各界的广泛关注.因此, 如何控制VOCs排放成为环境领域的热点.
低温等离子体通常被认为是固态、液态和气态以外的第四态, 当外加电压达到气体的着火电压时, 气体分子被击穿, 在这种状态下产生了自由基、各种离子、电子和原子的集合, 这些集合与VOCs发生碰撞, 将其转化成CO2、H2O或其他小分子物质(Whitehead, 2016).低温等离子体具有温度低、选择性高、操作方便、反应温和等优点, 是治理VOCs合适的选择(Thevenet et al., 2014).但低温等离子体技术还存在一些不可忽视的技术难题, 如能耗高、部分有机物质氧化不彻底、尾气中残留臭氧浓度过高等, 这些因素对低温等离子体技术的实践推广产生了一定的制约(Vandenbroucke et al., 2011).鉴于单独使用低温等离子体处理废气工艺中存在的问题, 国内外学者相继提出了低温等离子体协同催化氧化的耦合技术(Gong et al., 2018; Li et al., 2019; Shou et al., 2020).
锰基催化剂是一种活性高、稳定性强、价格低廉的非贵金属材料(王雪青等, 2019).研究表明, 低温等离子体协同锰基催化剂进行催化表现出反应条件温和、过程迅速和选择性高的优点(Qi et al., 2020), 可显著提高等离子体的能量利用效率, 减少副产物生成, 实现污染物的深度氧化.其中, LaMnO3催化剂作为贵金属催化剂在氧化还原过程中的潜在替代物之一, 因具有良好的催化活性和热稳定性而备受关注.OMS(KMn8O16 · nH2O)催化剂是一种以八面体MnO6为基础单元的锰氧化物, 可形成2×2隧道结构, 隧道内有K+进行电荷补偿.由于其独特的结构特点, OMS已被广泛研究并用于热催化氧化的各种应用.因此, 本研究在一系列锰基催化剂中选取LaMnO3、OMS(KMn8O16 · nH2O)两种催化剂进行制备, 考察其与等离子体协同催化乙酸乙酯在不同工况条件下的降解效率, 并对其降解效果进行比较与分析.
2 实验(Experiment part) 2.1 材料制备称取等物质的量的La(NO3)2 · 6H2O和Mn(NO3)2 · 4H2O溶解在适量蒸馏水中, 加入适量柠檬酸, 磁力搅拌下将溶液加热至80 ℃, 然后120 ℃干燥12 h, 随后在马弗炉中200 ℃(2 ℃ · min-1)处理1 h, 使用固态法制备LaMnO3催化剂(Lin et al., 2008).
称取9.48 g KMnO4和22.05 g Mn(Ac)2 · 4H2O在研钵中均匀研磨, 将粉末引入玻璃瓶中并在80 ℃下保持4 h, 得到黑色的粉末.洗涤后在100 ℃下干燥12 h, 使用溶胶-凝胶法制备OMS催化剂(Sihaib et al., 2017).
2.2 催化剂表征方法XRD采用德国布鲁克AXS公司生产的Bruker D5005型衍射仪对两种材料进行分析测定, 电压为40 kV, 电流为40 mA, 扫描范围为10°~80°.XPS在Thermo ESCALAB250XI系统上测定, 功率为150 W, 500 μm束斑, 处理软件为XPSPEAK.H2-TPR在具有TCD检测的商业BELCAT-B装置中进行, 将样品在Ar气流(50 cm3 · min-1)下250 ℃预处理30 min, 冷却后, 采用5%H2(体积分数)和Ar(50 cm3 · min-1)组成的气体混合物, 将温度从室温升至900 ℃(10 ℃ · min-1).BET使用Micromeritics Tristar 3000表面积和孔隙率分析仪测定, 测量之前, 样品在300 ℃下脱气3 h.SEM采用FEI Verios 460扫描电子显微镜对样品进行颗粒形状和颗粒粒径测量.TEM采用TF20型号透射电子显微镜对样品进行拍摄.
2.3 等离子体催化性能评价等离子体设备由进气系统、反应系统和检测仪器三部分组成(图 1).首先使空气通过干燥剂去除空气中的水分, 然后通过恒温水浴(40 ℃)挥发乙酸乙酯.通过控制质量流量控制器(MFC)的气流速率和乙酸乙酯的注入速率, 可以确定乙酸乙酯的进口浓度.在装有不同剂量催化剂的反应器中进行等离子体放电实验, 将催化剂包裹在石英棉中并置于放电间隙.等离子体反应器与之前使用的相同(Jiang et al., 2017), 由浙江大学制造.反应器由内径为1.75 cm、外径为2.61 cm、长度为39.8 cm的石英玻璃制成, 用长度为10 cm的铜片包裹并接地, 用直径为0.6 cm的不锈钢棒作为高压电极, 由南京苏曼制造的CTP-2000K为等离子体装置提供高压电源.采用杭州华南电子技术公司生产的DPO3012示波器测定放电电压.乙酸乙酯浓度通过FID气相色谱仪(Agilent 6890)测定.CO2浓度通过TCD气相色谱仪(Agilent 6890)测定.中间产物通过气相色谱-质谱联用仪(Agilent 7890A)测定.O3浓度通过泵吸式臭氧分析仪(XLA-BX-03)测定, 待等离子体出气稳定, 在出口处取样, 使用臭氧分析仪直接测量, 记录数据最大值.NOx浓度通过氮氧化物分析仪(Model 42i-HL)测定, 将等离子体排气管直接接入氮氧化物分析仪, 记录数据最大值.
 |
| 图 1 NTP处理VOCs的工艺流程图 Fig. 1 Schematic diagram of NTP for degradation of VOCs |
每次反应催化剂放置量为0.2 g, 反应条件为常温常压.乙酸乙酯降解率η和CO2选择性SCO2计算公式如下:
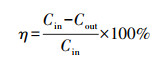
|
(1) |
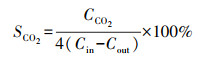
|
(2) |
式中, Cin和Cout分别为反应器进、出口处乙酸乙酯浓度(mg · m-3);CCO2为反应后产生的CO2浓度(mg · m-3).
3 结果与讨论(Results and discussion) 3.1 材料表征 3.1.1 SEM和TEM电镜表征图 2为LaMnO3、OMS的SEM图和TEM图.可以看出两种催化剂形态各异, LaMnO3催化剂颗粒比较规则, 呈现出棒状结构特性, 而OMS催化剂颗粒之间凝聚比较严重, 整体呈现出颗粒状态, 单位长度更小.TEM表征结果进一步证实了两种催化剂颗粒的差异, 从图中可以看出, LaMnO3催化剂粒径大约为400~500 nm, 而OMS催化剂粒径大约为100~200 nm.
 |
| 图 2 LaMnO3和OMS的SEM (A, C)和TEM (B, D) 图 Fig. 2 SEM (A, C) and TEM (B, D) images of LaMnO3 and OMS |
图 3为两种催化剂的XRD图谱.在OMS的图谱中, 主要出现的是MnO2(2θ=29°、38°)和Mn3O4(2θ=20°、22°和36°)的衍射峰, 由此判断Mn是以Mn4+和Mn3+的形式存在.LaMnO3样品的衍射峰与标准卡片#50-0299所对应的特征衍射峰完全一致, 由此可以判断制备的催化剂的晶型结构符合LaMnO3催化剂.OMS与LaMnO3相比具有更宽的衍射峰, 表明OMS具有更小的微晶尺寸.微晶尺寸越小, 比表面积越大, 有利于提高反应速率(Sun et al., 2011).
 |
| 图 3 各催化剂的XRD图谱 Fig. 3 XRD spectra of synthesized catalysts |
从表 1可以看出, OMS催化剂的比表面积和孔容明显比LaMnO3催化剂大.研究证明, 更大的比表面积可以提供更多的活性位点, 吸附更多的活性分子与目标污染物, 使污染物的停留时间更长, 增加污染物在等离子体中的接触, 因而具有更好的催化活性(梁文俊等, 2019).
| 表 1 不同催化剂的孔结构性质 Table 1 Pore structure properties of different catalysts |
图 4分别为LaMnO3、OMS的Mn 2p和O 1s的XPS图谱, 其中, Mn 2p在641.2(或641.8) eV处对应Mn3+, 在642.5(或643.4) eV处对应Mn4+.O 1s在529.8(或529.3) eV处对应晶格氧(Olatt), 在531.7 eV处对应吸附氧(Oads).从图中可以看出, OMS催化剂中Mn4+所对应的特征衍射峰比Mn3+更明显, 经过拟合得出, Mn4+/(Mn3++Mn4+)的比例为0.334, 明显优于LaMnO3的0.320, 因此, OMS催化剂比LaMnO3催化剂存在更多的Mn4+.同时, Oads的特征衍射峰也要比Olatt强.研究表明, Mn4+更有利O3分解, 从而产生更多活性氧原子, Oads反映了催化剂的氧迁移能力, Oads比例越高, 氧迁移能力越强, 更有利于催化剂利用活性氧原子.因此, Mn4+和Oads的含量越高, 越有利于对VOCs的去除(Wang et al., 2013; Song et al., 2018).
 |
| 图 4 各催化剂的XPS图谱 Fig. 4 XPS spectra of synthesized catalysts |
从图 5中的H2-TPR曲线可以观察到LaMnO3催化剂显示出两个还原峰, 这两个还原峰对应两步还原过程, 分别对应不同温度范围内(268 ℃为中心的峰宽和742 ℃为中心的强峰)Mn4+还原为Mn3+的过程.结果表明, Mn的两种氧化态(Mn4+和Mn3+)在初始催化剂中共存, OMS催化剂的还原曲线为单一强峰(264 ℃), 出现在比LaMnO3催化剂温度更低的地方, 表明OMS催化剂中的高价态锰离子在低温下比LaMnO3更易还原至低氧化态.
 |
| 图 5 各催化剂的H2-TPR图谱 Fig. 5 H2-TPR spectra of synthesized catalysts |
本研究选用LaMnO3和OMS两种催化剂耦合低温等离子体去除乙酸乙酯, 并在无催化剂条件下进行了对照实验.实验条件为:乙酸乙酯在放电区停留时间为1.5 s, 进气浓度为250 mg · m-3, 电压为4 kV, 催化剂放置量为0.2 g, 然后改变单一变量考察不同工况条件下对乙酸乙酯降解的影响.
3.2.1 输入电压的影响研究选用3、4、5和6 kV的电压结合催化剂考察对乙酸乙酯降解的影响.保持放电频率不变, 通过提升电压, 可以得到等离子体反应器的特定输入能量, 输入能量与电压提升的关系如图 6所示.从曲线的整体变化趋势可以看出, 特定的输入能量随电压的增大而增大, 随着电压从3 kV升至6 kV, 单独等离子体特定输入能量(SIE)从101.28 J · L-1升至193.27 J · L-1.一般来说, 电压升高, 电场强度升高, 可导致更多的微放电, 产生更多的活性物质用于降解乙酸乙酯, 从而提高乙酸乙酯的降解率.随着催化剂的加入, 去除效果显著提升, 在3 kV电压条件下, 分别加入LaMnO3和OMS, 乙酸乙酯去除率由63.1%分别升至73.9%和81.2%, OMS表现出更好的催化性能.因此, 对OMS进行分析, 可知电压由3 kV升至5 kV, 乙酸乙酯去除率由81.2%升至100%.原因是等离子体产生了更多的自由基、离子、电子和原子等活性物质, 活性物质的增加会增强与污染物的碰撞, 使污染物能更彻底的氧化, 从而提高了降解率.同时, 电压升高会去除吸附在催化剂表面的物质(白文文等, 2020), 恢复催化剂的活性, 也间接提高了去除率.
 |
| 图 6 不同电压对放电能量和乙酸乙酯降解效率的影响 Fig. 6 The influence of different voltages on discharge energy and degradation efficiency of ethyl acetate |
分别考察进气浓度为250、350、450和550 mg · m-3条件下等离子体耦合催化剂对乙酸乙酯降解的影响(图 7a).随着LaMnO3和OMS两种催化剂的加入, 乙酸乙酯去除效率提升明显, 在250 mg · m-3进气浓度条件下, 去除率由87.2%分别上升到91.0%和95.6%, OMS催化剂表现出更好的降解性能.因此, 进一步考察在OMS催化剂条件下进气浓度由250 mg · m-3升至550 mg · m-3时乙酸乙酯去除率的变化, 结果显示去除率从95.6%降至69.8%.这一方面是因为进气浓度增加, 乙酸乙酯分子数目增多, 等离子体产生的活性物质和污染物碰撞不充分, 造成污染物氧化不彻底;另一方面是因为进气浓度增加导致催化剂活性位点饱和(Shahna et al., 2016), 减少了污染物在等离子体中的停留时间, 从而降低了去除率.
 |
| 图 7 初始进气浓度(a)、不同停留时间(b) 及不同催化剂放置量(c) 对乙酸乙酯降解效率的影响 Fig. 7 Effect of different initial concentrations (a), residence time(b) and catalyst placement (c) on the degradation efficiency of ethyl acetate |
等离子体选用1.0、1.5和2.0 s的停留时间为变量协同催化剂考察对乙酸乙酯降解的影响(图 7b).在停留时间为1.0 s的条件下, 单独等离子体的去除率为68.9%, 加入LaMnO3和OMS催化剂后去除率分别达到74.1%和81.5%, OMS催化剂表现出比LaMnO3催化剂更好的催化活性.选用OMS催化剂考察在不同停留时间下的去除效率, 当停留时间由1.0 s变为2.0 s时, 乙酸乙酯的降解率由81.5%升至84.8%.原因是随着停留时间的延长, 乙酸乙酯能吸收更多的活性物质, 使活性物质与污染物发生的碰撞更充分, 单位体积内形成的物质更活跃(Devahasdin et al., 2003), 同时也增大了污染物与催化剂的接触时间, 使氧化更彻底, 从而提高了降解率.
3.2.4 催化剂放置量的影响分别选用0.1、0.2、0.3和0.4 g催化剂放置量考察对乙酸乙酯降解的影响(图 7c).当催化剂放置量为0.1 g时, LaMnO3和OMS两种催化剂对乙酸乙酯的去除率分别为83.3%和89.7%, 可见OMS催化剂具有更好的催化性能.但随着催化剂放置量的增大, 乙酸乙酯去除率先升高后降低, 出现该现象的原因是催化剂增加一方面提高了Mn4+和Oads的数量, 使污染物能更好地与催化剂接触, 增加了污染物在等离子体中的停留时间, 使去除率上升;另一方面, 等离子体的放电模式随催化剂放置量的改变而改变, 在同一电压下, 等离子体放电产生的自由基和活性物质数量是相似的, 随着催化剂的增多会影响正常放电, 等离子体所产生的高能电子和自由基等物质减少, 导致低温等离子体的放电效果减弱进而影响去除效率.
3.3 降解乙酸乙酯产物的分析研究选用LaMnO3和OMS两种催化剂耦合低温等离子体去除乙酸乙酯, 并在无催化剂条件下进行了对照实验.实验条件为:乙酸乙酯在放电区停留时间为1.5 s, 进气浓度为250 mg · m-3, 通过改变电压对以下产物进行分析.
3.3.1 CO2选择性由图 8可知, 在低温等离子体协同催化剂降解体系中, 随着催化剂的加入和电压的升高, 反应产物中CO2的选择性提高.原因是催化剂的加入使反应体系中出现了大量氧空位和吸附氧物种等活性物质, 产生的活性物质使乙酸乙酯氧化更彻底, 从而有效提高了CO2的形成.电压的升高有利于高能量电子和活性物质的产生, 有利于乙酸乙酯气体与高能电子碰撞产生的各种氧化物质的过程(Allah et al., 2014), 从而进一步加速乙酸乙酯氧化成CO2的过程.
 |
| 图 8 不同电压对CO2选择性的影响 Fig. 8 The influence of different voltages on CO2 selectivity |
低温等离子体技术对乙酸乙酯的去除效果显著, 但该技术还存在一些不可忽视的问题, 如有机物质氧化不彻底, 以及尾气中存在O3和NOx等污染物质, 通过等离子体耦合催化剂和适当提高电压可以明显改善上述情况.如图 9所示, 催化剂的加入可明显降低O3的产生量, 原因是产生的O3和催化剂发生催化分解, 将臭氧分解为O2等物质.随着电压的升高O3产生量先明显增长然后趋于平缓, 原因是电压升高可产生更多的高能电子, 将氧气分解成活性氧原子(式(1)), 然后氧气和氧原子碰撞聚合生成O3(式(2)) (Pekarek, 2003), 因此, O3浓度呈上升趋势;随着放电电压和能量效率的提高, 生成的O3进一步与部分脱附的活性氧原子发生反应(Huang et al., 2011), 参与污染物降解, 同时系统放电所产生的热量使得反应器温度上升, 部分O3受热分解.
 |
| 图 9 不同电压对O3和NOx产生量的影响 Fig. 9 The influence of different voltages on the amount of O3 and NOx produced |
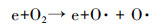
|
(1) |
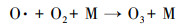
|
(2) |
NOx产生量增加是因为随着电压升高, 等离子体放电区域内产生了更多的高能电子、N ·和O · 自由基, N ·和O · 反应会生成NO, 产生的NO会被臭氧产生的活性氧原子和等离子体产生的活性物质氧化成NO2(Chang et al., 2019), 由此产生更多NOx.随着催化剂的加入, NOx产生量则随着催化剂的加入明显降低, 原因是催化剂的加入可以将NOx直接分解, 生成N2和O2, 其中, OMS催化剂的作用明显优于LaMnO3催化剂.
3.3.3 其它降解产物低温等离子体耦合催化剂处理乙酸乙酯过程中, 乙酸乙酯并未完全降解, 因此, 利用气相色谱-质谱联用仪(GC-MS)对反应副产物进行分析.在进气浓度为250 mg · m-3, 停留时间为2 s, 电压为4 kV的条件下, 分别对NTP、NTP+LaMnO3、NTP+OMS的尾气采用活性炭管吸收0.5 h, 然后将吸附尾气的活性炭放入甲醇中解吸1 h, 最后通过GC-MS分析中间副产物.在低温等离子体催化降解乙酸乙酯体系中, 产生了大量高能电子、活性自由基等物质, 这些活性物质都可以激发乙酸乙酯, 促进乙酸乙酯的降解.由图 10及表 2可知, 单独等离子体去除乙酸乙酯, 反应产物中生成了甲醛、乙醛、乙醇、3-戊烯、1-己醇等小分子物质, 等离子体催化体系加入LaMnO3和OMS催化剂后, 这些小分子物质进一步氧化, 生成了甲醛、庚烷、十一烷等物质, 这说明催化剂的加入可以有效去除中间副产物的产生, 使乙酸乙酯降解更完全.
 |
| 图 10 乙酸乙酯中间产物质谱图 Fig. 10 The mass spectrums of Ethyl acetate intermediate |
| 表 2 乙酸乙酯中间产物汇总 Table 2 Ethyl acetate intermediate product summary table |
选用有较高催化活性的OMS催化剂考察其长期运行的稳定性(图 11), 实验条件为:乙酸乙酯进气浓度为250 mg · m-3, 气体停留时间为1.5 s, 放电电压为3 kV和5 kV交替.打开等离子体反应器, 持续进行催化降解实验, 每隔2 h测定乙酸乙酯的去除率.每12 h进行电压交替, 目的是通过提升电压去除吸附在催化剂表面占据活性位点的物质, 恢复催化剂活性, 使催化剂具有良好的耐久性.经过2个循环后, 乙酸乙酯的去除率和CO2选择性都保持稳定, 说明OMS催化剂耦合低温等离子体催化降解乙酸乙酯可以长时间保持稳定, 并且可以通过提升电压的方法恢复催化剂的活性.
 |
| 图 11 OMS催化剂结合等离子体去除乙酸乙酯稳定性 Fig. 11 OMS catalyst combined with plasma to remove ethyl acetate Stability |
1) 通过XPS、XRD、BET、H2-TPR、SEM和TEM特性表征可知, OMS催化剂比表面积、吸附氧、Mn4+均强于LaMnO3催化剂, 说明OMS催化剂催化性能更优.
2) 采用OMS、LaMnO3催化剂耦合低温等离子体催化降解乙酸乙酯, 发现在不同工况下污染物去除率随污染物初始浓度的增大而减小, 随停留时间增长而变大, 当催化剂放置量为0.2 g时达到最佳催化效果.
3) 催化剂的加入明显减少了O3的生成, OMS催化剂比LaMnO3催化剂的效果要强.随着电压增大, NOx产生量增大, 但催化剂的加入能有效减少NOx的生成.
4) 在等离子体去除乙酸乙酯催化体系中, OMS催化剂在两个电压循环期间保持了极高的催化活性, 说明OMS催化剂耦合低温等离子体催化降解乙酸乙酯可以长时间保持稳定, 并通过提高电压恢复活性.
5) 采用气相色谱-质谱联用仪(GC-MS)对反应副产物进行分析, 发现催化剂的加入可有效抑制中间副产物的出现, OMS催化剂的抑制效果最佳.
致谢(Acknowledgement):: 感谢浙江工业大学环境学院宋爽教授对论文英文摘要的修改.
Allah Z A, Whitehead J C, Martin P. 2014. Remediation of dichloromethane (CH2Cl2) using non-thermal, atmospheric pressure plasma generated in a packed-bed reactor[J]. Environmental Science & Technology, 48(1): 558-565. |
白文文, 秦彩虹, 郑洋, 等. 2020. 介质阻挡放电联合锰基催化剂对乙酸乙酯的降解效果[J]. 环境工程学报, 14(5): 1294-1303. |
Chang T, Lu J, Shen Z, et al. 2019. Post plasma catalysis for the removal of acetaldehyde using Mn-Co/HZSM-5 Catalysts[J]. Industrial & Engineering Chemistry Research, 58(32): 14719-14728. |
Devahasdin S, Fan C, Li K, et al. 2003. TiO2 photocatalytic oxidation of nitric oxide: Transient behavior and reaction kinetics[J]. Journal of Photochemistry & Photobiology A Chemistry, 156(1/3): 161-170. |
Gong X, Zhao R, Qin J, et al. 2018. Ultra-efficient removal of NO in a MOFs-NTP synergistic process at ambient temperature[J]. Chemical Engineering Journal, 358: 291-298. |
Huang H, Ye D, Leung D Y C, et al. 2011. Byproducts and pathways of toluene destruction via plasma-catalysis[J]. Journal of Molecular Catalysis A Chemical, 336(1/2): 87-93. |
Jiang L, Nie G, Zhu R, et al. 2017. Efficient degradation of chlorobenzene in a non-thermal plasma catalytic reactor supported on CeO2/HZSM-5 catalysts[J]. Journal of Environmental Ences, 55(5): 266-273. |
Li K, Ji J, Huang H, et al. 2019. Efficient activation of Pd/CeO2 catalyst by non-thermal plasma for complete oxidation of indoor formaldehyde at room temperature[J]. Chemosphere, 246: 125762. |
梁文俊, 孙慧频, 朱玉雪, 等. 2019. 流向变换等离子体催化系统去除甲苯[J]. 中国环境科学, 39(12): 4974-4981. |
Lin T, Zhang Q, Li W, et al. 2008. Monolith manganese-based catalyst supported on ZrO2-TiO2 for NH3-SCR reaction at low temperature[J]. Acta Physico-Chimica Sinica, 24(7): 1127-1131. DOI:10.1016/S1872-1508(08)60046-7 |
Pekarek S. 2003. Non-Thermal Plasma Ozone Generation[J]. Acta Polytechnica, 43(6): 47-51. |
Qi Y, Shan X, Wang M, et al. 2020. Study on low-temperature scr denitration mechanisms of manganese-based catalysts with different carriers[J]. Water Air and Soil Pollution, 231(6): 437-443. |
Shahna F G, Bahrami A, Alimohammadi I, et al. 2016. Chlorobenzene degeradation by non-thermal plasma combined with EG-TiO2/ZnO as a photocatalyst: Effect of photocatalyst on CO2 selectivity and byproducts reduction[J]. Journal of Hazardous Materials, 324: 544-553. |
Shou T, Li Y, Bernards M T, et al. 2020. Degradation of gas-phase O-xylene via combined non-thermal plasma and Fe doped LaMnO3 catalysts: Byproduct control[J]. Journal of Hazardous Materials, 387: 121750. DOI:10.1016/j.jhazmat.2019.121750 |
Sihaib Z, Puleo F, Garcia-Vargas J M, et al. 2017. Manganese oxide-based catalysts for toluene oxidation[J]. Applied Catalysis B: Environmental, 209: 689-700. DOI:10.1016/j.apcatb.2017.03.042 |
Song H, Hu F, Peng Y, et al. 2018. Non-thermal plasma catalysis for chlorobenzene removal over CoMn/TiO2 and CeMn/TiO2: synergistic effect of chemical catalysis and dielectric constant[J]. Chemical Engineering Journal, 347: 447-454. DOI:10.1016/j.cej.2018.04.018 |
Subrahmanyam C, Renken A, Kiwi-Minsker L. 2010. Catalytic non-thermal plasma reactor for abatement of toluene[J]. Chemical Engineering Journal, 160(2): 677-682. DOI:10.1016/j.cej.2010.04.011 |
Sun H, Chen S, Wang P, et al. 2011. Catalytic oxidation of toluene over manganese oxide octahedral molecular sieves (OMS-2) synthesized by different methods[J]. Chemical Engineering Journal, 178: 191-196. DOI:10.1016/j.cej.2011.10.047 |
Thevenet F, Sivachandiran L, Guaitella O, et al. 2014. Plasma-catalyst coupling for volatile organic compound removal and indoor air treatment: A review[J]. Journal of Physics D: Applied Physics, 47(22): 224011. DOI:10.1088/0022-3727/47/22/224011 |
Vandenbroucke A M, Rino M, De Geyter Nathalie, et al. 2011. Non-thermal plasmas for non-catalytic and catalytic VOC abatement[J]. Journal of Hazardous Materials, 195: 30-54. DOI:10.1016/j.jhazmat.2011.08.060 |
Wang Z, Shen G, Li J, et al. 2013. Catalytic removal of benzene over CeO2-MnOx composite oxides prepared by hydrothermal method[J]. Applied Catalysis B: Environmental, 138: 253-259. |
Whitehead J C. 2016. Plasma-catalysis: the known knowns, the known unknowns and the unknown unknowns[J]. Journal of Physics D: Applied Physics, 49(24): 243001. DOI:10.1088/0022-3727/49/24/243001 |
王雪青, 黎欢毅, 王邦芬, 等. 2019. 等离子体场内CeO2催化降解甲醇的表面活性氧物种来源与作用研究[J]. 环境科学学报, 39(8): 2725-2734. |