 2021, Vol. 41
2021, Vol. 41



2. 中国科学院生态环境研究中心, 中国科学院饮用水科学与技术重点实验室, 北京 100085;
3. 天津市大港镀锌厂, 天津 300270
2. Key Laboratory of Drinking Water Science and Technology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085;
3. Tianjin Dagang Galvanizing Factory, Tianjin 300270
硝酸盐是水中常见的污染物, 由于工业生产中大量使用硝酸等化学药剂, 导致工业废水中硝酸盐浓度较高, 若随意排放会造成严重的硝酸盐污染.硝酸盐污染对环境及人体都会造成不利影响, 一方面过量的氮进入水体会造成水体富营养化, 加剧水体污染;另一方面, 水环境的污染会对饮用水安全造成威胁, 硝酸盐被摄入到人体后易被还原细菌还原为亚硝酸盐, 亚硝酸盐可将血红蛋白的亚铁离子氧化为三价铁离子, 阻碍红细胞的氧气转运, 从而导致紫绀、脑缺氧甚至死亡(Sergi et al., 2018).同时, 亚硝酸盐在胃中与氨氮结合形成的亚硝基氨化合物具有致癌的作用(Archna et al., 2015;Chai et al., 2017).因此, 水中的硝酸盐污染问题已经引起人们的广泛关注.
目前常见的硝酸盐去除方法主要有生物法(范彬等, 2000;Zhang et al., 2010;姚静华等, 2012;于海彤等, 2019)、物理化学法(Liou et al., 2006;Zhang et al., 2012)和电化学还原法(Zhang et al., 2018).其中, 生物法是目前最常用的硝酸盐去除方法, 具有成本低、应用广泛等优点, 但也存在运行周期长、受外界环境因素(如温度等)影响大的问题.物理化学方法如反渗透、电渗析和离子交换(Kalaruban et al., 2016)等, 对进水水质的要求较高, 且存在浓缩废水的二次排放问题.电化学技术由于无需或少需额外投加药剂、反应副产物容易控制、占地面积少、可实现自动化控制等优势, 受到了越来越多的关注(曲久辉等, 2007).
电还原硝酸盐的还原路径和最终产物主要由电极材料决定(Dash et al., 2005).在电化学还原硝酸盐研究中, 前期往往通过将贵金属如Pt(Rudnev et al., 2009)、Pd(Pizarro et al., 2018)和Au(El-Deab, 2004)等负载材料作为阴极实现对硝酸盐的高效还原, 但由于成本原因难以进行大规模的推广和应用.近年来, 研究人员尝试用一些非贵金属材料代替贵金属修饰电极还原硝酸盐.例如, 刘蕾等(2013)以碳纳米管修饰的石墨阴极还原硝酸盐, 证实了碳材料对硝酸盐具有较高的还原效率.Li等(2009b)对Cu、Fe和Ti阴极在相同反应条件下还原硝酸盐的效率进行研究发现, Fe相比于Cu和Ti作阴极对硝酸盐的还原效率更高.
本研究通过筛选采用铁板为阴极, Ti/RuO2电极为阳极, 构建电化学同步还原/氧化连续流反应体系, 并针对电镀锌硝酸酸洗废水进行电化学脱氮研究, 考察电流密度和水力停留时间对硝酸盐及总氮去除效率的影响, 并在此基础上对体系的稳定性进行考察.
2 实验部分(Experimental section) 2.1 实验试剂实验装置如图 1所示, 连续流反应装置由DH1765-1高精度直流稳压电源、亚克力电解槽、阴极、阳极、MS-H-Pro+型磁力搅拌器和L100-1S-2型蠕动泵组成.反应槽为长方体构型, 有效容积为230 mL.阴极为4块135 mm×45 mm×1.5 mm的铁板, 阳极为2块与阴极极板同尺寸的Ti/RuO2金属网, 单块极板有效反应面积为81 cm2.硝酸钾(KNO3)、氯化钠(NaCl)、硫酸钠(Na2SO4)、氢氧化钠(NaOH)和硫酸(H2SO4)等药品购于国药化学试剂公司, 叔丁醇(C4H10O)购于阿拉丁试剂公司, 所有药品纯度均为分析纯.
 |
| 图 1 实验装置示意图 Fig. 1 Schematic diagram of electrochemical divice |
实验前将铁板在稀硝酸中浸泡10 min, 取出后用砂纸进行充分打磨, 使其表面灰黑色氧化物完全去除, 呈现光亮的银灰色.所用废水为取自某镀锌厂的电镀锌硝酸酸洗废水, 其中, 锌离子浓度为679 mg·L-1, 因此, 采用调节原水pH的方式对原水进行预处理.经过预处理后的废水水质见表 1.
| 表 1 预处理后的废水水质情况 Table 1 Water quality after pretreatment |
取1000 mL经过预处理的废水, 除有特殊说明外均按照3 g·L-1的剂量投加NaCl, 搅拌均匀后加入废水池中, 将电解槽与废水池通过蠕动泵连接, 废水通过蠕动泵输入电解槽中.以铁板为阴极, Ti/RuO2电极为阳极, 通过改变直流电源的输出电流控制极板的电流密度, 通过调节蠕动泵流速控制水力停留时间, 通电后定时取样并分别检测样品中的NO3--N、NH4+-N、NO2--N和TN的浓度.
2.4 分析方法硝酸盐浓度的测定采用紫外分光光度法(HJ/T 346—2007), 总氮浓度的测定采用碱性过硫酸钾消解-紫外分光光度法(HJ 636—2012), 亚硝酸盐浓度的测定采用分光光度法(GB/T 7493—1987), 氨氮浓度的测定采用纳氏试剂分光光度法(HJ 535—2009), 锌离子浓度和总铁浓度采用Z-2000原子吸收分光光度计(日立仪器有限公司)检测.电极表面形貌在ZEISS MERLIN Compact型扫描电子显微镜上观察, 同时配备X-MAX-20能谱分析仪(英国牛津仪器公司).
3 结果与讨论(Results and discussion) 3.1 阴极材料对脱氮效能的影响选取了实际工程中常用的钛板及不锈钢板作为阴极, 与铁板的脱氮效果进行对比.实验结果如图 2所示, 出水中主要含氮污染物为硝酸盐, 其中, 铁板作阴极时出水硝酸盐浓度低于50 mg·L-1, 而不锈钢与钛板作阴极时出水硝酸盐浓度分别为100和110 mg·L-1左右.在相同反应条件下铁板表现出了明显优于钛板和不锈钢的脱氮性能, 与相关文献(Dash et al., 2005;Li et al., 2009b)的报道一致, 因此, 在后续试验中阴极选择铁板.铁阴极脱氮效果优于钛板和不锈钢的原因可通过反应过程中电极电压的不同解释.Talhi等(2011)发现不锈钢阴极在高反应电压时, 由于析氢副反应的存在出现硝酸盐还原效率随电压升高而降低的现象.研究表明, 铁与钛作阴极时析氢电位(-1.0 ~-1.2 V)几乎一致(Li et al., 2009b), 而不锈钢的析氢电位(-0.9 V)(张海兵等, 2016)略正于铁和钛.本组实验在电流密度为7.5 mA·cm-2时以钛板和不锈钢作阴极时的反应电压(4.082 V和3.870 V)高于铁板(3.607 V), 此时钛阴极和不锈钢阴极由于具有更负的阴极电位而产生更强的析氢反应, H+占据更多活性位点从而降低了硝酸盐的还原效率.
 |
| 图 2 阴极材料对出水硝酸盐(a)、氨氮(b)、亚硝酸盐(c)和总氮(d)浓度的影响 (电流密度7.5 mA·cm-2, NaCl投加量3 g·L-1, 水力停留时间3 h) Fig. 2 Effect of cathode material on the concentration of nitrate(a), ammonia(b), nitrite(c) and total nitrogen(d) in effluent |
考察了连续流体系在不同电流密度下各含氮污染物的稳定出水浓度.如图 3所示, 水力停留时间为3 h, 电流密度的升高对硝酸盐还原效率有明显的提升.原因是电流密度的升高会加快电子的转移, 提高污染物的去除效率.此外, 电流密度由1.5 mA·cm-2提高到6 mA·cm-2, 出水氨氮浓度均较高, 电流密度提高到7.5 mA·cm-2时出水中氨氮浓度大幅降低.分析原因, 当电流密度升高时, 施加到阳极的电压会同时升高(Kim et al., 2006), 而阳极电势是影响活性氯转化效率的关键因素.Li等(2019)研究发现, 在1.5 V和2.0 V的阳极电势下观察到的氨氮转化率较低, 当阳极电势提高为2.5 V时会发生氨氮的快速转化, 原因是阳极电势高于氯离子氧化所需的电势(约2.3 V)(Vecitis et al.2011).本实验中电流密度由6.0 mA·cm-2升高到7.5 mA·cm-2时, 如图 3b所示, 电压由3.269 V升至3.607 V, 在这一过程中施加到阳极的实际电势可能超过了Cl-氧化所需的电势, 导致Cl-在阳极被快速氧化为活性氯, 从而大幅提高了体系中氨氮的氧化效率.
 |
| 图 3 电流密度对出水硝酸盐(a)、氨氮(b)、亚硝酸盐(c)和总氮(d)浓度的影响 (NaCl投加量3 g·L-1, 水力停留时间3 h) Fig. 3 Effect of current density on the concentration of nitrate(a), ammonia(b), nitrite(c) and total nitrogen (d) in effluent |
研究了不同水力停留时间对连续流脱氮体系脱氮性能的影响.如图 4所示, 电流密度为7.5 mA·cm-2, 水力停留时间由2 h延长至3 h时, 出水中的总氮浓度由95 mg·L-1降为50 mg·L-1.原因是当水力停留时间延长时, 废水中的硝酸盐与阴极的接触时间延长, 还原更充分, 同时硝酸盐氮还原产生的氨氮副产物也可以被活性氯充分氧化从而降低出水浓度, 由此推断水力停留时间的进一步延长将获得更低的出水污染物浓度, 但会明显增加单位能耗.因此, 为兼顾处理效果和经济性, 在后续处理环节有效降低污染负荷的同时控制成本, 设置水力停留时间为3 h.
 |
| 图 4 水力停留时间对出水硝酸盐(a)、氨氮(b)、亚硝酸盐(c)和总氮(d)浓度的影响 (电流密度7.5 mA·cm-2, NaCl投加量3 g·L-1) Fig. 4 Effect of HRT on the concentration of nitrate (a), ammonia(b), nitrite(c) and total nitrogen(d) in effluent |
实验所用废水为某镀锌厂的电镀锌酸洗废水, 具体水质见表 1.反应器连续运行发现, 随着运行时间的延长, 体系的脱氮效率逐渐下降.为解决阴极还原效率下降的问题, 每7 d为一个周期, 一个周期结束后将阴极极板取出酸洗并用砂纸清理阴极表面后继续运行, 连续运行21 d.如图 5所示, 对阴极极板进行上述处理可使体系恢复初始脱氮效能, 运行过程中出水氨氮与亚硝酸盐浓度均低于1 mg·L-1, 硝酸盐浓度与总氮浓度相近, N2选择性高于99%.
 |
| 图 5 连续流反应器中硝酸盐、氨氮、亚硝酸盐和总氮的出水浓度 (电流密度7.5 mA·cm-2, NaCl投加量3 g·L-1, 水力停留时间3 h) Fig. 5 The effluent concentration of nitrate, ammonia, nitrite and total nitrogen in the continuous flow reactor |
对每个周期结束后经过处理的极板进行称重计算阴极极板的损耗, 结果如表 2所示, 连续运行7 d并进行酸洗和使用砂纸对阴极表面进行清理后, 阴极极板的损耗率最高仅有0.42%, 表现出了铁板作为阴极具有较好的耐久性.
| 表 2 阴极极板损耗 Table 2 Loss of cathode plate |
利用扫描电子显微镜对反应前后的阴极进行了表征.图 6a为经过酸洗处理后的阴极极板反应前的形貌, 表面较光滑, 无附着物, 仅有部分砂纸清理造成的条形痕迹.图 6b为反应后的阴极形貌, 极板表面大面积覆盖着沉积物, 进一步放大可以看到沉积物为颗粒状物质且较密集.如图 6c、6d所示, 对比反应前后阴极的EDS能谱图发现, 反应后阴极表面的Fe元素比例明显减少, O元素比例大幅上升, 且出现了反应前阴极表面不存在的Ca、Mg和Zn元素.由此可以推断, 电解过程中废水中存在的部分Ca2+、Mg2+和Zn2+沉积在阴极, 同时随着运行时间的延长阴极表面的Fe逐渐被氧化, 导致阴极表面活性位点减少和导电能力下降, 从而造成硝酸盐还原效率降低和电压升高.
 |
| 图 6 反应前(a)和反应后(b)阴极的SEM图及反应前(c)和反应后(d)阴极的EDS能谱图 Fig. 6 SEM images of the cathodebefore (a) and after(b) the reaction and the EDS spectrum of the cathode before(c) and after(d) the reaction |
在溶液中存在Cl-时, 硝酸盐的主要还原产物氨氮会与阳极氧化产生的活性氯继续反应生成N2.为更准确地核算阴极电流效率, 在电流密度为7.5 mA·cm-2、水力停留时间为3 h和不投加NaCl的条件下对目标废水进行电催化脱氮处理.实验结果显示, 出水硝酸盐浓度为110.24 mg·L-1, 还原产物为氨氮、亚硝酸盐和N2等气体, 所占比例分别为65.5%、0.6%和33.9%.Wang等(2006)针对电还原硝酸盐的气体产物进行了分析, 结果显示气体产物主要为N2, 而NO和NO2等气体产物的量极低, 因此, 在本文计算中近似认为硝酸盐还原的气体产物为N2.阴极电流效率采用式(1)计算(Georgeaud et al., 2011; Ghazouani et al., 2016).

|
(1) |
式中, Ci和Ct分别为进水和出水硝酸盐浓度(mol·L-1);n为生成1 mol产物所需电子转移数(当反应产物为NH4+、N2和NO2-时, n分别取8、5、3);F为法拉第常数(取值为96485 C·mol-1);Vr为反应器有效容积(L);I为反应电流(A);t为电解时间(s), 此处取10800 s.经核算, 在电流密度7.5 mA·cm-2和水力停留时间3 h的条件下, 阴极脱氮电流效率为12.5%.
3.6 脱氮机理分析硝酸盐电催化还原主要有两种方式, 一种是间接还原(李熔等, 2016), 溶液中的H+在阴极被还原为H*, NO3-被阴极表面吸附的H*还原;另一种是直接还原(叶舒帆等, 2011;Xu et al., 2018), 即NO3-在阴极直接被电子还原.有研究表明, 叔丁醇可作为原子H*的淬灭剂将H*转变为惰性自由基(Liu et al., 2017), 为研究H*在硝酸盐还原过程中的作用, 在不同叔丁醇浓度条件下考察硝酸盐的还原效率.如图 7所示, 随着叔丁醇浓度由0 mmol·L-1升高到2 mmol·L-1, 硝酸盐的还原速率出现了小幅度的下降, 继续增加叔丁醇浓度, 硝酸盐的还原速率没有明显的变化.由此可见, 铁阴极还原硝酸盐的过程中直接还原起主要作用, 具体反应过程见式(2)~(6)(叶舒帆等, 2011;Xu et al., 2018).
 |
| 图 7 不同浓度叔丁醇对硝酸盐还原效率的影响 (NO3--N初始浓度50 mg·L-1, 电流密度7.5 mA·cm-2, Na2SO4浓度50 mmol·L-1) Fig. 7 Effects of different concentrations of tert-butanol on nitrate reduction efficiency |
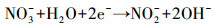
|
(2) |
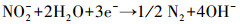
|
(3) |
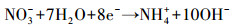
|
(4) |
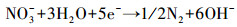
|
(5) |
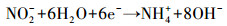
|
(6) |
如图 8所示, 在不投加NaCl时体系中硝酸盐浓度随反应时间的延长逐渐下降, 氨氮浓度逐渐升高, 在投加2 g·L-1 NaCl时, 反应过程中几乎无副产物氨氮的累积.由此可见, 氨氮和氮气等气体是铁阴极还原硝酸盐的主要产物, 溶液中Cl-的存在是氨氮快速氧化去除的主要原因, 与相关文献(Su et al., 2017;Li et al., 2009a)的研究结果一致.氨氮氧化去除的主要作用机制为溶液中的Cl-在阳极被氧化为具有较强氧化性的活性氯, 活性氯与溶液中的氨氮反应生成N2, 具体反应过程见式(7)~(9)(Rao et al., 2019).
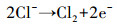
|
(7) |
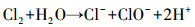
|
(8) |
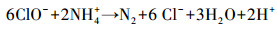
|
(9) |
 |
| 图 8 NaCl投加量为0 g·L-1(a)和2 g·L-1(b)时硝酸盐、氨氮和亚硝酸盐的浓度变化 (NO3--N初始浓度70 mg·L-1, 电流密度7.5 mA·cm-2, Na2SO4浓度10 mmol·L-1) Fig. 8 Variation in the concentration of nitrate, ammonia and nitrite at NaCl dosage of 0 g·L-1(a) and 2 g·L-1(b) |
综上所述, N元素在体系中的去除是阴极直接还原和阳极间接氧化协同作用的结果.溶液中的硝酸盐在阴极通过电子的直接还原作用被还原为氨氮、N2等气体和少量的亚硝酸盐, 溶液中Cl-在阳极的作用下被氧化为ClO-, ClO-可高选择性地将溶液中的氨氮氧化为N2, 从而实现体系中总氮的降低.
4 结论(Conclusions)本研究构建了连续流同步电还原/氧化脱氮体系, 在电流密度7.5 mA·cm-2和水力停留时间3 h条件下可将出水硝酸盐浓度由初始的410.7 mg·L-1降低至45.0 mg·L-1, 出水总氮浓度由初始的421.6 mg·L-1降至约50.0 mg·L-1.提高电流密度和延长水力停留时间有利于提高硝酸盐的去除效率.通过考察体系的稳定性发现, 由于阴极表面的氧化和溶液中金属离子的沉积, 体系脱氮效率随反应时间的延长逐渐降低, 但阴极极板经酸洗后可恢复初始脱氮效率.机理研究发现, 体系中N元素去除的主要机制是阴极直接还原和阳极间接氧化的协同作用.
Archna, Sharma S K, Sobti R C. 2015. Nitrate removal from ground water: A review[J]. Journal of Chemistry, 9(4): 1667-1675. |
Chai L Y, Peng C, Min X B, et al. 2017. Two-sectional struvite formation process for enhanced treatment of copper-ammonia complex wastewater[J]. Transactions of Nonferrous Metals Society of China, 27(2): 457-466. DOI:10.1016/S1003-6326(17)60052-9 |
Dash B P, Chaudhari S. 2005. Electrochemical denitrificaton of simulated ground water[J]. Water Research, 39(17): 4065-4072. DOI:10.1016/j.watres.2005.07.032 |
El-Deab M S. 2004. Electrochemical reduction of nitrate to ammonia at modified gold electrodes[J]. Electrochimica Acta, 49(9/10): 1639-1645. |
范彬, 曲久辉, 刘锁祥, 等. 2000. 饮用水中硝酸盐的脱除[J]. 环境污染治理技术与设备, 1(3): 44-50. |
Georgeaud V, Diamand A, Borrut D, et al. 2011. Electrochemical treatment of wastewater polluted by nitrate: selective reduction to N2 on boron-doped diamond cathode.[J]. Water Science and Technology, 63(2): 206-212. DOI:10.2166/wst.2011.034 |
Ghazouani M, Akrout H, Jomaa S, et al. 2016. Enhancing removal of nitrates from highly concentrated synthetic wastewaters using bipolar Si/BDD cell: Optimization and mechanism study[J]. Journal of Electroanalytical Chemistry, 783: 28-40. DOI:10.1016/j.jelechem.2016.10.048 |
Kalaruban M, Loganathan P, Shim W G, et al. 2016. Removing nitrate from water using iron-modified Dowex 21K XLT ion exchange resin: Batch and fluidised-bed adsorption studies[J]. Separation and Purification Technology, 158: 62-70. DOI:10.1016/j.seppur.2015.12.022 |
Kim K W, Kim Y J, Kim I T, et al. 2006. Electrochemical conversion characteristics of ammonia to nitrogen[J]. Water Research, 40(7): 1431-1441. DOI:10.1016/j.watres.2006.01.042 |
Liou Y H, Lo S L, Kuan W H, et al. 2006. Effect of precursor concentration on the characteristics of nanoscale zerovalent iron and its reactivity of nitrate[J]. Water Research, 40(13): 2485-2492. DOI:10.1016/j.watres.2006.04.048 |
Li F, Peng X, Liu Y, et al. 2019. A chloride-radical-mediated electrochemical filtration system for rapid and effective transformation of ammonia to nitrogen[J]. Chemosphere, 229: 383-391. DOI:10.1016/j.chemosphere.2019.04.180 |
Li L, Liu Y. 2009a. Ammonia removal in electrochemical oxidation: Mechanism and pseudo-kinetics[J]. Journal of Hazardous Materials, 161(2/3): 1010-1016. |
Li M, Feng C, Zhang Z, et al. 2009b. Efficient electrochemical reduction of nitrate to nitrogen using Ti/IrO2-Pt anode and different cathodes[J]. Electrochimica Acta, 54(20): 4600-4606. DOI:10.1016/j.electacta.2009.03.064 |
李熔, 宋长忠, 赵旭, 等. 2016. Pd/rGO/C电极催化还原硝酸盐[J]. 环境工程学报, 10(2): 648-654. |
刘蕾, 张冬梅, 褚衍洋. 2013. 电化学法去除水中的硝酸根[J]. 环境工程学报, 7(11): 4195-4200. |
Liu Y Z, Mao R, Tong Y T, et al. 2017. Reductive dechlorination of trichloroacetic acid (TCAA) by electrochemical process over Pd-In/Al2O3 catalyst[J]. Electrochimica Acta, 232: 13-21. DOI:10.1016/j.electacta.2017.02.071 |
Pizarro A H, Torija I, Monsalvo V M. 2018. Enhancement of Pd-based catalysts for the removal of nitrite and nitrate from water[J]. Journal of Water Supply: Research and Technology-Aqua, 67(7): 615-625. |
曲久辉, 刘会娟. 2007. 水处理电化学原理与技术[M]. 北京: 科学出版社.
|
Rao X, Shao X, Xu J, et al. 2019. Efficient nitrate removal from water using selected cathodes and Ti/PbO2 anode: Experimental study and mechanism verification[J]. Separation and Purification Technology, 216: 158-165. DOI:10.1016/j.seppur.2019.02.009 |
Rudnev A V, Molodkina E B, Ehrenburg M R, et al. 2009. Methodical aspects of studying the electroreduction of nitrate on modified single crystal Pt(hkl)+Cu electrodes[J]. Russian Journal of Electrochemistry, 45(9): 1052-1063. DOI:10.1134/S1023193509090110 |
Sergi G S, Mariana L L, Kiril H, et al. 2018. Electrocatalytic reduction of nitrate: Fundamentals to full-scale water treatment applications[J]. Applied Catalysis B: Environmental, 236: 546-568. DOI:10.1016/j.apcatb.2018.05.041 |
Su L, Li K, Zhang H, et al. 2017. Electrochemical nitrate reduction by using a novel Co3O4/Ti cathode[J]. Water Research, 120: 1-11. DOI:10.1016/j.watres.2017.04.069 |
Talhi B, Monette F, Azzouz A. 2011. Effective and selective nitrate electroreduction into nitrogen through synergistic parameter interactions[J]. Electrochimica Acta, 58: 276-284. DOI:10.1016/j.electacta.2011.09.044 |
Vecitis C D, Schnoor M H, Rahaman M S, et al. 2011. Electrochemical multiwalled carbon nanotube filter for viral and bacterial removal and inactivation[J]. Environmental Science & Technology, 45(8): 3672-9. |
Wang Y, Qu J H, Wu R C, et al. 2006. The electrocatalytic reduction of nitrate in water on Pd/Sn-modified activated carbon fiber electrode[J]. Water Research, 40(6): 1224-1232. DOI:10.1016/j.watres.2006.01.017 |
Xu D, Li Y, Yin L, et al. 2018. Electrochemical removal of nitrate in industrial wastewater[J]. Frontiers of Environmental Science & Engineering, 12(3): 1-1. |
姚静华, 赵国智, 田光明, 等. 2012. 复三维电极-生物膜反应器脱除饮用水中硝酸盐的试验研究[J]. 环境科学学报, 32(6): 1333-1341. |
叶舒帆, 胡筱敏, 和英滇, 等. 2011. 非贵金属催化还原水中的硝酸盐氮[J]. 环境化学, 30(10): 1711-1717. |
于海彤, 白雪, 董琪, 等. 2019. 废水中对甲酚、硝酸盐和氨氮同步去除效能研究[J]. 环境科学学报, 39(10): 3279-3283. |
Zhang B, Sun B, Ji M, et al. 2010. Quantification and comparison of ammonia-oxidizing bacterial communities in MBRs treating various types of wastewater[J]. Bioresource Technology, 101(9): 3054-3059. DOI:10.1016/j.biortech.2009.12.048 |
张海兵, 马力, 闫永贵, 等. 2016. 17-4PH不锈钢的析氢行为[J]. 腐蚀与防护, 37(4): 317-320. |
Zhang L J, Tao H C, Wei X Y, et al. 2012. Bioelectrochemical recovery of ammonia-copper(Ⅱ) complexes from wastewater using a dual chamber microbial fuel cell[J]. Chemosphere, 89(10): 1177-1182. DOI:10.1016/j.chemosphere.2012.08.011 |
Zhang Y, Li J, Bai J, et al. 2018. Exhaustive conversion of inorganic nitrogen to nitrogengas based on a photoelectro-chlorine cycle reaction and a highly selective nitrogen gas generation cathode[J]. Environmental Science and Technology, 52(3): 1413-1420. DOI:10.1021/acs.est.7b04626 |