 2021, Vol. 41
2021, Vol. 41



2. 中国科学院安徽光学精密机械研究所大气物理化学实验室, 合肥 230031
2. Laboratory of Atmospheric Physico-Chemistry, Anhui Institute of Optics and Fine Mechanics, Chinese Academy of Sciences, Hefei 230031
人为源排放的苯、甲苯等苯系物是大气挥发性有机物的组成部分, 也是危害性较大的环境污染物(Li et al., 2020;Yousefian et al., 2020). 苯是最简单的苯系物, 它具有一定的毒性和致癌性(Garg et al., 2019;Ji et al., 2020;Yousefian et al., 2020). 同时苯会与OH自由基发生光氧化反应, 形成半挥发性和难挥发性产物, 这些产物通过凝结或气/粒分配后形成二次有机气溶胶(Secondary Organic Aerosol, SOA) (Peng et al., 2017;Al-Naiema et al., 2020). SOA粒子主要是粒径小于2.5 μm的细粒子(PM2.5), 它们可以深入到肺部和支气管, 严重危害人类健康(Chowdhury et al., 2018;Chowdhury et al., 2019). 此外, SOA粒子还是城市光化学烟雾的主要组分, 它们能够吸收、散射太阳光, 降低大气能见度, 是形成灰霾天气的元凶, 已逐渐成为影响辐射强迫、扰动区域气候变化的重要驱动因子(Baltensperger, 2010;Moise et al., 2015). 随着气候变化影响的加剧, SOA光学特性的研究已成为大气化学领域的热点.
农业生产和汽车尾气排放的氨与SO2在水汽存在下反应产生的硫酸铵((NH4)2SO4)是大气中常见的无机气溶胶细粒子(Shi et al., 2017). (NH4)2SO4细粒子其大的比表面积可作为半挥发化合物凝结和非均相反应中心, 进而影响SOA的化学组分和理化性质(Tajuelo et al., 2019). 早期的烟雾箱实验结果表明, 低浓度(NH4)2SO4细粒子(0.2~100 μg·m-3)虽然不会影响苯系物SOA的产率, 但能够提高SOA的形成速率(Cocker et al., 2001;Hao et al., 2007). 高浓度(NH4)2SO4细粒子(大于100 μg·m-3)能够提供更多的反应位点, 有利于半挥发产物在其比表面凝结. 此外, (NH4)2SO4细粒子表面吸湿后产生氢离子和铵根离子, 氢离子能够催化二醛化合物发生水合、聚合等非均相反应形成高分子量产物(Lu et al., 2009), 而铵离子能与二醛化合物发生亲核加合、脱水、成环等反应产生咪唑类化合物. 这些产物具有较低的挥发性, 容易进入颗粒相, 从而提高SOA的产率(Lu et al., 2009;Huang et al., 2016). 产生的咪唑类化合物含有CN双键等生色团, 具有一定的吸光能力, 是大气棕色碳的主要组分(Laskin et al., 2015;Grace et al., 2020).
人们常用质量吸收系数(mass absorption coefficient, MAC)来表征气溶胶粒子的光学特性. 利用滤膜采样器采集气溶胶样品, 用水或甲醇等溶剂超声提取颗粒物中可溶性组分, 借助紫外-可见分光光度计等宽波段光谱仪测量光通过提取液后的光衰减, 得到吸光度, 根据吸收池的长度和样品中有机物的浓度, 获得MAC参数(Updyke et al., 2012;Powelson et al., 2014;Moise et al., 2015):
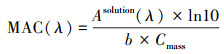
|
(1) |
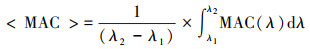
|
(2) |
式中, Asolution为提取液在特定波长处的吸光度, ln10为常数, b为光程长度, Cmass为提取液中SOA有机碳的质量浓度. 根据式(2)在测量波长范围内(λ1-λ2)积分得到平均MAC值< MAC>. Updyke等(2012)测得氨存在时1, 3, 5-三甲苯和α-蒎烯等SOA粒子在500 nm处的 < MAC>值为10~1000 cm2·g-1, 300~700 nm内 < MAC>的值约为300 cm2·g-1. 尽管上述烟雾箱实验(Cocker et al., 2001;Hao et al., 2007;Lu et al., 2009;Huang et al., 2016)开展了(NH4)2SO4细粒子对苯系物SOA产率的影响, 但对SOA的MAC等光学特性的影响情况则未见报道.
随着我国城市化过程的加快, 大气中含有浓度较高的(NH4)2SO4细粒子. 如北京和上海等特大城市发生雾霾污染时, 气溶胶颗粒中H+、NH4+、SO42-和其他无机离子的浓度可达100~300 μg·m-3 (Wang et al., 2017;Shao et al., 2018). 课题组前期利用气溶胶激光飞行时间质谱仪测量(NH4)2SO4细粒子存在时苯系物SOA的化学组分, 获得了咪唑、咪唑醛等咪唑类产物(Huang et al., 2016;Huang et al., 2017;张威等, 2020). 在此基础上, 本文利用烟雾箱系统在不同浓度、酸度和相对湿度下的(NH4)2SO4细粒子条件下, 启动苯光氧化产生SOA粒子, 利用离线滤膜-溶剂提取-连续光谱法测量苯SOA粒子在200~600 nm内的平均MAC值< MAC>, 获得不同浓度、酸度和湿度的(NH4)2SO4细粒子对苯SOA的MAC影响规律, 为研究高浓度(NH4)2SO4细粒子背景下人为源SOA的光学特性提供实验依据.
2 材料与方法(Materials and methods) 2.1 药品苯(> 99%)购自汕头市达濠精细化学品有限公司;甲醇(> 99%)购于Sigma Aldrich(上海)贸易有限公司;硫酸铵(> 99.0%)、硫酸(98%)、氢氧化钠(> 99%)购自国药集团化学试剂有限公司. 上述药品未经处理直接用于实验.
2.2 烟雾箱实验实验在400 L烟雾箱装置中进行. 该装置主要由清洁空气产生系统、进样系统、光照反应系统和收集检测系统组成(徐俊等, 2018;张威等, 2020). 采用数显温湿度计(WSB-2, 郑州博洋仪器仪表有限公司)测量箱内的温湿度. 所有实验箱内温度为(25±1) ℃, 除相对湿度实验外, 箱内相对湿度(RH)为37%±3%. 空气压缩机(ACO-012, 森森集团股份有限公司)产生的压缩空气通过变色硅胶、活性炭和Balston DFU Grade BX过滤器得到清洁空气. 用PM2.5粒子检测仪(CEM-DT96, 深圳华盛昌机械实业有限公司)和泵吸式VOCs检测仪(XLA-BX-VOC, 深圳市普利通电子科技有限公式)测得清洁空气中颗粒物和挥发性有机物的浓度均为零, 满足实验要求. 每次实验前先用200 L清洁空气清洗Teflon气袋3次, 再向气袋充入约1/3体积清洁空气, 用微升注射器抽取一定量苯注入到加热至80 ℃的液体气化瓶中, 使其挥发完全, 并随清洁空气通入烟雾箱. 臭氧由臭氧发生器(XM-TS, 青岛新美净化设备有限公司)产生, 通入烟雾箱一定时间, 随后用清洁空气充至满体积. 用泵吸式苯检测仪(Ⅱ 2G Baseefa, 英国离子科学(中国)有限公司), 臭氧分析仪(SPT-GT-1000-O3, 北京思普特科技有限公司)测得烟雾箱内苯和臭氧的体积比浓度为1×10-6和10×10-6. 将4 g·L-1 (NH4)2SO4溶液用气溶胶发生器(TSI 9302, 美国TSI公司)雾化产生粒径小于2 μm的细粒子, 再通过硅胶干燥管后通入烟雾箱. 用PM2.5粒子检测仪实时测量箱内细粒子的浓度, 当测得的浓度达到设定浓度后, 立即停止通入(NH4)2SO4细粒子. 配气结束后, 随即打开4只特征波长为254 nm的紫外灯光照, 臭氧在254 nm的紫外光照射下产生OH自由基, 启动苯发生光氧化反应. 采用泵吸式苯检测仪、臭氧分析仪和PM2.5粒子检测仪测量烟雾箱内苯、臭氧和颗粒物浓度(徐俊等, 2018;张威等, 2020).
由于本实验使用的烟雾箱体积较小, 气溶胶粒子由于湍流, 布朗扩散和重力沉降等沉积在烟雾箱壁上. 为了确定气溶胶粒子在壁上的损耗速率, 参照Takekawa等(2003)和课题组前期的表征方法(Huang et al., 2013), 用TSI 9302气溶胶发生器雾化0.04 g·L-1(NH4)2SO4溶液, 通过硅胶干燥管后进入烟雾箱内一定时间后, 用清洁空气充至满体积. 采用扫描电迁移率粒径谱仪(TSI 3080L DMA, TSI 3775 CPC, 美国TSI公司)测量不同时间下不同粒径(NH4)2SO4细粒子的数量浓度, 研究其衰减情况. 实验平行进行3次, 取其平均值进行分析.
2.3 实验方法所有实验烟雾箱内苯和臭氧体积比浓度固定控制为1×10-6和10×10-6. (NH4)2SO4细粒子浓度实验中, 通过调节细粒子通入时间来控制其在烟雾箱内的浓度, 使用PM2.5粒子检测仪监测箱内细粒子浓度, 依次设定为25、50、75、100、200、300、400、500 μg·m-3. 相对湿度实验中, 箱内(NH4)2SO4细粒子的浓度固定设为100 μg·m-3, 调节清洁空气通过装有超纯水洗瓶的时间来控制烟雾箱内相对湿度, 使用温湿度计监测箱内相对湿度, 使其分别为40%、50%、60%、70%和80%. 根据Wang等(2019)的外场测量结果, 大气中颗粒物一般呈酸性, pH低于4. 但受到沙尘和颗粒物组分二次反应的影响, 颗粒物pH值呈现弱碱性, 大气颗粒物pH约为1.1~8.4. 因此, 在(NH4)2SO4细粒子酸度实验中, 分别使用0.1 mol·L-1的H2SO4和NaOH溶液将(NH4)2SO4溶液pH值调为1、3、5、7和8, 产生细粒子后通入箱内, 浓度控制为100 μg·m-3. 每个实验平行3次, 光照反应4 h后, 将烟雾箱内的颗粒物以4 L·min-1的流速收集到聚四氟乙烯滤膜上, PM2.5粒子检测仪测得通过滤膜后的气体颗粒物浓度为零, 表明SOA粒子被收集到滤膜上. 随后将滤膜放置于5 mL的2%甲醇中进行2次超声提取, 每次超声30 min. 以2%甲醇溶液为参比, 第二次超声提取液用双光束紫外-可见分光光度计(UV-6100s, 上海Mapada公司)进行测量, 测得的吸收光谱近似为直线, 表明SOA在第一次超声时已全部溶于2%甲醇溶液. 采用紫外-可见分光光度计和总有机碳分析仪(TOC-L, 日本Shimadzu公司)测量第一次提取液在200~600 nm的吸收光谱和有机碳总浓度. 将测得的有机碳总浓度减去2%甲醇溶液的有机碳浓度, 获得提取的苯SOA有机碳浓度. 根据式(1)和(2)计算得到苯SOA在200~600 nm内的 < MAC>值.
3 结果与讨论(Results and discussion) 3.1 (NH4)2SO4细粒子对苯SOA形成和吸光产物的影响气溶胶粒子的壁效应损耗可以用一阶指数变化描述, 损耗大小依赖于损耗系数kdep(dp):

|
(3) |
式中, N(dp)和kdep(dp)分别为粒径为dp的气溶胶数浓度和一阶损耗系数(Takekawa et al., 2003). 测得的气溶胶壁损耗速率系数kdep(dp)与气溶胶粒径dp的关系如图 1所示. 根据Takekawa等(2003)的实验结果, kdep(dp)与dp具有如下关系:
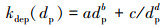
|
(4) |
 |
| 图 1 气溶胶壁损耗速率系数kdep与气溶胶粒径dp的关系图 Fig. 1 The deposition rate efficient (kdep) of the particles as the function of particle diameter(dp) |
利用式(4)对实验数据进行非线性最小二乘拟合得到参数a、b、c、d分别为5.89×10-13、6.04、12.47和1.16. 对于粒径为300 nm的气溶胶粒子, kdep等于30.5%·h-1, 大于Takekawa等(2003)搭建的烟雾箱的kdep值(10.3%·h-1). 这主要是因为本文使用的烟雾箱体积(400 L)比Takekawa等(2003)的烟雾箱体积(2000 L)小, 气溶胶粒子的壁效应较大.
前期的烟雾箱实验证实, OH自由基与苯主要发生加合反应, OH自由基添加到苯环上形成羟基环己二烯基自由基, 由图 2可知, 该自由基与氧气分子反应产生过氧自由基, 过氧自由基经过一系列反应, 芳香环裂解产生乙二醛、2-丁烯二醛、2, 3-环氧-丁二醛等羰基产物(Sato et al., 2012;Wang et al., 2013;Wang et al., 2016). 这些产物在达到其饱和蒸气压后通过自身成核凝结形成SOA粒子. 因此, 在光照的前15 min内, 烟雾箱内SOA粒子经壁效应矫正后的浓度仅为12 μg·m-3. 如图 3所示, 随着光照反应的进行, 形成更多的羰基产物参与气相/粒子相均分, SOA的浓度逐渐增大. 反应180 min时, 泵吸式苯检测仪未能探测到苯, 而箱内SOA经壁效应矫正后的浓度达到最大值233 μg·m-3. 随后, 由于苯反应完全, 光照反应趋于停止, SOA粒子的浓度经壁效应矫正后几乎保持不变(Huang et al., 2016;徐俊等, 2018;张威等, 2020).没有(NH4)2SO4细粒子存在时, 1×10-6苯和10×10-6臭氧光氧化反应产生的SOA粒子的紫外-可见吸收光谱在205 nm处存在羰基化合物特征吸收峰(Carlton et al., 2007)证实这些实验结果(图 4). 羰基产物仅包含C=C和C=O双键, 不包含强生色团和助色团(Laskin et al., 2015;Moise et al., 2015), 因而粒子具有较弱的吸光能力.
 |
| 图 2 硫酸铵细粒子存在时苯光氧化反应产生咪唑类有机物的可能机理 Fig. 2 The possible formation mechanism of imidazole compounds formed from the photooxidation of benzene in the presence of ammonium sulfate fine particle |
 |
| 图 3 没有和存在100 μg·m-3硫酸铵细粒子时不同光照时间下经壁效应矫正后的苯SOA浓度 Fig. 3 The wall effect corrected concentration of benzene SOA in the absence and presence of 100 μg·m-3 ammonium sulfate fine particle at different irradiation time |
 |
| 图 4 没有和存在100 μg·m-3硫酸铵细粒子时苯SOA不同波长下的质量吸收系数 Fig. 4 The mass absorption coefficient of benzene SOA at different wavelengths in the absence and presence of 100 μg·m-3 ammonium sulfate fine particle |
对于(NH4)2SO4细粒子实验, 从测得的粒子浓度减去(NH4)2SO4细粒子初始浓度, 得到苯SOA浓度. 100 μg·m-3 (NH4)2SO4细粒子存在时, 如图 3所示, 苯SOA浓度随光照反应时间的变化曲线与没有细粒子的情形相类似. 然而, 光照180 min时, 烟雾箱内苯SOA经壁效应矫正后最大浓度达到352 μg·m-3, 比没有细粒子的情形增加了51%. 而图 4显示的100 μg·m-3 (NH4)2SO4细粒子存在时, 苯SOA的紫外-可见吸收曲线与没有细粒子的明显不同, 最大的吸收峰红移至280 nm. 根据Maxut等(2015)的实验结果, 该吸收峰为咪唑类化合物分子C=N双键中的n→π*跃迁产生的特征吸收峰, 这表明咪唑类化合物是(NH4)2SO4细粒子存在时苯SOA的主要组分. 当1×10-6苯和10×10-6臭氧反应体系中存在(NH4)2SO4细粒子时, OH自由基氧化苯形成的乙二醛等气相羰基产物在(NH4)2SO4细粒子表面凝结形成SOA粒子. 如图 2所示, 乙二醛等气相羰基产物在(NH4)2SO4细粒子表面发生水合形成四醇产物, 还与NH4+反应形成二胺产物, 四醇与二胺中间产物通过亲核进攻、脱水、成环等系列反应产生咪唑、咪唑醛等含氮有机物(Maxut et al., 2015;Huang et al., 2016). SOA中C=N生色团的增加增强了吸光能力. 100 μg·m-3 (NH4)2SO4细粒子存在时, 测得的200~600 nm内苯SOA的 < MAC>值为316 cm2·g-1, 与Updyke等(2012)测得的氨老化的SOA粒子的 < MAC>值(300 cm2·g-1)较为接近, 比没有细粒子存在时苯SOA的 < MAC>值(225 cm2·g-1)增加了40%. 这表明, (NH4)2SO4细粒子能够影响 < MAC>等光学参数, 可以通过苯SOA粒子的MAC变化情况来分析(NH4)2SO4细粒子对苯系物SOA光学特性影响规律.
3.2 不同浓度(NH4)2SO4细粒子对苯SOA光学特性的影响(NH4)2SO4细粒子比表面是苯光氧化产生的半挥发性和难挥发性产物凝结和非均相反应中心. (NH4)2SO4细粒子浓度的大小决定了其比表面的大小, 从而影响咪唑类产物的形成. 测得的不同浓度的(NH4)2SO4细粒子存在下, 苯SOA粒子的MAC值见图 5. 当硫酸铵细粒子浓度低于100 μg·m-3时, 苯SOA粒子的MAC值为228~232 cm2·g-1, 吸光能力弱. 如图 6所示, 当硫酸铵细粒子浓度低于100 μg·m-3时, 箱内苯SOA经壁效应矫正后的最大浓度约为237~243 μg·m-3, 与没有细粒子的情形相近. 这些苯SOA粒子的紫外-可见吸收光谱只在205 nm处存在羰基化合物特征吸收峰, 没有探测到280 nm处的吸收峰. 图 7显示了不同浓度硫酸铵细粒子存在时苯SOA提取液在205 nm和280 nm处的吸光度. 当硫酸铵细粒子浓度低于100 μg·m-3时, 苯SOA提取液在205 nm处的吸光度为0.957~0.972, 与没有细粒子时的吸光度0.984相近. 而提取液在280 nm处的吸光度接近为零, 表明苯SOA中几乎没有咪唑类化合物, 羰基化合物是苯SOA的主要组分. 这些表明, 低浓度的(NH4)2SO4细粒子(小于100 μg·m-3)仅作为苯SOA凝结的核心, 不会影响苯SOA的产率和颗粒相产物(Cocker Ⅲ et al., 2001;Hao et al., 2007).
 |
| 图 5 不同浓度硫酸铵细粒子存在时苯SOA平均质量吸收系数值 Fig. 5 Averaged mass absorption coefficient of benzene SOA in the presence of different concentrations of ammonium sulfate fine particle |
 |
| 图 6 不同浓度硫酸铵细粒子存在时经壁效应矫正后的最大苯SOA浓度 Fig. 6 The wall effect corrected maximum concentration of benzene SOA in the presence of different concentrations of ammonium sulfate fine particle |
 |
| 图 7 不同浓度硫酸铵细粒子存在时苯SOA提取液在205 nm和280 nm处的吸光度 Fig. 7 The absorbance of 205 nm and 280 nm of the extract solution of benzene SOA in the presence of different concentrations of ammonium sulfate fine particle |
当(NH4)2SO4细粒子浓度高于100 μg·m-3时, 苯SOA粒子MAC值(图 5)和经壁效应矫正后的浓度(图 6)显著增加, 并随着(NH4)2SO4细粒子浓度的增大而逐渐增大. 如图 7所示的苯SOA提取液在205 nm吸光度显著减少, 并随着(NH4)2SO4细粒子浓度的增大而逐渐减小, 而在280 nm处的吸光度则呈现相反的规律. 这证实随着(NH4)2SO4细粒子浓度的增大, 更多乙二醛等羰基化合物参与反应产生咪唑类化合物. 当(NH4)2SO4细粒子浓度超过300 μg·m-3后, 苯SOA粒子的浓度和光学性质趋于稳定, 提取液在205 nm和280 nm处的吸光度几乎保持不变. 根据Ahlberg等(2019)和Lu等(2009)的实验结果, 高浓度(NH4)2SO4细粒子具有较大的比表面积, 能够为非均相反应提供更多反应位点. 此外, 苯系物光氧化产生的乙二醛等产物在高浓度(NH4)2SO4细粒子表面凝结, 单位比表面积上凝结的乙二醛等产物变少, 使得乙二醛与铵离子的反应更完全, 形成更多的颗粒相咪唑类产物, 导致SOA粒子浓度增大, 粒子吸光能力逐渐增强. 由于实验1×10-6苯和10 ×10-6臭氧固定不变, 反应形成的乙二醛等产物是一定的. 当(NH4)2SO4细粒子浓度增大至一定值时(如300 μg·m-3), 将大部分的二醛产物反应产生咪唑类化合物. 此后, (NH4)2SO4细粒子浓度继续增大, 咪唑类化合物将不再显著增大. 因而苯SOA粒子的浓度、组分和光学性质趋于稳定.
3.3 不同相对湿度下(NH4)2SO4细粒子对苯SOA光学特性的影响水分子能够促进细粒子的吸湿, 有利于乙二醛水合反应的发生和NH4+的产生, 从而影响SOA的组分和光学特性(Tajuelo et al., 2019). 在100 μg·m-3 (NH4)2SO4细粒子存在时, 相对湿度从40%增大至80%时, 测得的苯SOA粒子的MAC值见图 8a. 当相对湿度低于70%时, 苯SOA粒子的MAC值随着相对湿度的增加而逐渐增大. 当相对湿度为70%时, 苯SOA粒子的MAC值达到最大值378 cm2·g-1. 但是当相对湿度高于70%时, 苯SOA粒子的MAC值则随着相对湿度的增加而逐渐减小.
 |
| 图 8 100 μg·m-3硫酸铵细粒子存在时, 不同相对湿度下苯SOA平均质量吸收系数值(a)及苯SOA提取液在205 nm和280 nm处的吸光度(b) Fig. 8 Averaged mass absorption coefficient (a) and the absorbance of 205 nm and 280 nm of the extract solution of benzene SOA(b) at different relative humidity in the presence of 100 μg·m-3 ammonium sulfate fine particle |
随着相对湿度的增大, (NH4)2SO4细粒子表面吸湿后产生更多的NH4+, 有利于乙二醛等羰基化合物在其表面发生非均相反应, 导致苯SOA中乙二醛等羰基化合物的含量减少, 苯SOA提取液在205 nm处的吸光度随着相对湿度的增加而逐渐减小(图 8b). 由图 2所示的反应机理可知, 乙二醛产物在(NH4)2SO4细粒子表面发生的与NH4+反应和水合反应是存在竞争的. 在相对湿度小于70%时, (NH4)2SO4细粒子表面吸湿后主要产生NH4+, 主要发生NH4+与乙二醛反应, 形成咪唑类吸光产物(Maxut et al., 2015;Fan et al., 2020). 因此随着相对湿度的增大, 苯SOA粒子的吸光能力逐渐增强(图 8a), 提取液在280 nm处的吸光度逐渐增大(图 8b). 而相对湿度高于70%时, (NH4)2SO4细粒子表面吸湿后含有较多的水分, 除了产生NH4+外, OH自由基启动苯光氧化产生的有机酸产物也会溶解或吸附在(NH4)2SO4细粒子表面而产生H+, H+催化乙二醛水合反应增强, 较多的四醇产物能够发生脱水聚合形成高分子量产物(Gomez et al., 2015). 这导致NH4+与乙二醛反应被部分减弱, 咪唑类吸光产物减少. 高分子量产物的含量随着相对湿度的增大而逐渐增多. 相比于咪唑类化合物, 形成的高分子量产物没有生色团和助色团, 吸光能力弱, 使得苯SOA粒子的MAC值(图 8a)和提取液在280 nm处的吸光度(图 8b)随着相对湿度的增大而减小.
3.4 不同酸度(NH4)2SO4细粒子对苯SOA光学特性的影响大气中不同来源的(NH4)2SO4细粒子具有不同的酸度. 本实验配制系列pH值溶液产生细粒子模拟不同酸度的(NH4)2SO4细粒子. 测得的不同pH溶液产生的100 μg·m-3 (NH4)2SO4细粒子存在时, 苯SOA粒子的MAC值如图 9a所示. 从图中可知, 苯SOA粒子的MAC值随着(NH4)2SO4溶液pH值的增大而增加. 当pH值为8时, SOA粒子的MAC值达到最大值347 cm2·g-1. 而当pH=1时, 苯SOA粒子的MAC值最小(224 cm2·g-1), 表明强酸性的(NH4)2SO4细粒子不利于咪唑类产物的产生. 这主要是因为强酸性的(NH4)2SO4细粒子表面含有大量H+, 催化大部分乙二醛等羰基化合物发生水合等反应, 使得苯SOA中乙二醛等羰基化合物的含量骤减, 如图 9b所示的苯SOA提取液在205 nm的吸光度最小. 水合反应产生的四醇主要发生脱水聚合形成吸光能力较弱的高分子量产物(Gomez et al., 2015), 因而苯SOA提取液咪唑类化合物含量少, 在280 nm处的吸光度小. 随着pH值的增加, H+减少, 被催化的乙二醛等羰基化合物随之减少, SOA中乙二醛等羰基化合物的含量增加, 提取液在205 nm的吸光度随pH值的增加而逐渐增大.而随着pH值的增大, NH4+反应增强, 形成的咪唑类化合物增多, 苯SOA粒子的MAC值(图 9a)和提取液在280 nm的吸光度(图 9b)逐渐增大.
 |
| 图 9 不同pH值溶液产生的100 μg·m-3硫酸铵细粒子存在时苯SOA平均质量吸收系数值(a)及苯SOA提取液在205 nm和280 nm处的吸光度(b) Fig. 9 Averaged mass absorption coefficient (a) and the absorbance of 205 nm and 280 nm of the extract solution of benzene SOA in the presence of 100 μg·m-3 ammonium sulfate fine particle produced by solution with different pH value |
相反的, 在弱碱性(NH4)2SO4细粒子存在下, 由于碱的潮解特性, (NH4)2SO4细粒子吸湿性增强, 颗粒表面含有较多的水分, 除了产生较多NH4+外, OH-还会与NH4+反应产生具有较强亲核能力的NH3, 促进更多的乙二醛等羰基化合物参与反应, 产生更多的二胺中间产物, 从而导致苯SOA中的羰基组分减少, 提取液在205 nm的吸光度减小(图 9b). 二胺中间产物进一步反应产生咪唑类化合物, 从而导致苯SOA中咪唑类产物含量增多(Kampf et al., 2012;De Haan et al., 2017), MAC值(图 9a)和提取液在280 nm的吸光度(图 9b)增大. 因此, 当大气中的(NH4)2SO4细粒子受到沙尘等影响呈现弱碱性时, 更有利于咪唑类化合物的形成, 可能会促进灰霾污染的产生.
4 结论(Conclusions)城市大气中常见的(NH4)2SO4细粒子大的比表面积能够成为苯系物光氧化产生的乙二醛等气相产物凝结和反应中心, 促进咪唑类吸光产物的形成, 从而影响SOA的光学特性. 苯SOA在200~600 nm内的平均质量吸收系数值随着(NH4)2SO4细粒子浓度和相对湿度的增大而逐渐增大. 但当相对湿度高于70%时, 乙二醛等产物的水合反应增强, 吸光能力较弱的高分子量产物增多, 苯SOA的MAC值反而减小. 强酸性的(NH4)2SO4细粒子不利于咪唑类产物的产生, 苯SOA的MAC值随着(NH4)2SO4细粒子酸度的降低而逐渐增大. 而弱碱性(NH4)2SO4细粒子吸湿后产生亲核能力更强的NH3, 形成的咪唑类产物增多, MAC值增大. 然而本实验采用离线方法检测SOA的MAC值, 后续实验可开展SOA消光与散射系数等光学参数在线测量研究.
Ahlberg E, Eriksson A, Brune W H, et al. 2019. Effect of salt seed particle surface area, composition and phase on secondary organic aerosol mass yields in oxidation flow reactors[J]. Atmospheric Chemistry and Physics, 19(4): 2701-2712. DOI:10.5194/acp-19-2701-2019 |
Al-Naiema I M, Offenberg J H, Madler C J, et al. 2020. Secondary organic aerosols from aromatic hydrocarbons and their contribution to fine particulate matter in Atlanta, Georgia[J]. Atmospheric Environment, 223(117227): 1-9. |
Baltensperger U. 2010. Aerosols in clearer focus[J]. Science, 329(5998): 1468-1470. |
Carlton A G, Turpin B J, Altieri K E, et al. 2007. Atmospheric oxalic acid and SOA production from glyoxal: Results of aqueous photooxidation experiments[J]. Atmospheric Environment, 41(35): 7588-7602. DOI:10.1016/j.atmosenv.2007.05.035 |
Chowdhury P H, He Q F, Male T L, et al. 2018. Exposure of lung epithelial cells to photochemically aged secondary organic aerosol shows increased toxic effects[J]. Environmental Science & Technology Letters, 5(7): 424-430. |
Chowdhury P C, He Q F, Carmieli R, et al. 2019. Connecting the oxidative potential of secondary organic aerosols with reactive oxygen species in exposed lung cells[J]. Environmental Science & Technology, 53(23): 13949-13958. |
Cocker Ⅲ D R, Clegg S L, Flagan R C, et al. 2001. The effect of water on gas-particle partitioning of secondary organic aerosol. Part Ⅱ. m-xylene and 1, 3, 5-trimethylbenzene photooxidation systems[J]. Atmospheric Environment, 35(35): 6073-6085. DOI:10.1016/S1352-2310(01)00405-8 |
De Haan D O, Hawkins L N, Welsh H G, et al. 2017. Brown carbon production in ammonium- or amine-containing aerosol particles by reactive uptake of methylglyoxal and photolytic cloud cycling[J]. Environmental Science & Technology, 51(13): 7458-7466. |
Fan M J, Ma S Q, Ferdousi N, et al. 2020. Modeling of carbonyl/ammonium sulfate aqueous brown carbon chemistry via UV/Vis spectral decomposition[J]. Atmosphere, 11(358): 1-15. |
Garg A, Gupta N C. 2019. A comprehensive study on spatio-temporal distribution, health risk assessment and ozone formation potential of BTEX emissions in ambient air of Delhi, India[J]. Science of the Total Environment, 659: 1090-1099. DOI:10.1016/j.scitotenv.2018.12.426 |
Gomez M E, Lin Y, Guo S, et al. 2015. Heterogeneous chemistry of glyoxal on acidic solutions. An oligomerization pathway for secondary organic aerosol formation[J]. The Journal of Physical Chemistry A, 119(19): 4457-4463. DOI:10.1021/jp509916r |
Grace D N, Lugos E N, Ma S Q, et al. 2020. Brown carbon formation potential of the biacetyl- ammonium sulfate reaction system[J]. ACS Earth and Space Chemistry, 4(7): 1104-1113. DOI:10.1021/acsearthspacechem.0c00096 |
Hao L Q, Wang Z Y, Huang M Q, et al. 2007. Effects of seed aerosols on the growth of secondary organic aerosols from the photooxidation of toluene[J]. Journal of Environmental Sciences, 19(6): 704-708. DOI:10.1016/S1001-0742(07)60117-X |
Huang M Q, Hao L Q, Gu X J, et al. 2013. Effects of inorganic seed aerosols on the growth and chemical composition of secondary organic aerosol formed from OH-initiated oxidation of toluene[J]. Journal of Atmospheric Chemistry, 70(2): 151-164. DOI:10.1007/s10874-013-9262-9 |
Huang M Q, Zhang J H, Cai S Y, et al. 2016. Mass spectrometric study of aged benzene secondary organic aerosol in the presence of dry ammonium sulfate[J]. Journal of Atmospheric Chemistry, 73(3): 329-344. DOI:10.1007/s10874-016-9328-6 |
Huang M Q, Hao L Q, Cai S Y, et al. 2017. Effects of inorganic seed aerosols on the particulate products of aged 1, 3, 5-trimethylbenzene secondary organic aerosol[J]. Atmospheric Environment, 152: 490-502. DOI:10.1016/j.atmosenv.2017.01.010 |
Ji Y Y, Gao F H, Wu Z H, et al. 2020. A review of atmospheric benzene homologues in China: Characterization, health risk assessment, source identification and countermeasures[J]. Journal of Environmental Sciences, 95: 225-239. DOI:10.1016/j.jes.2020.03.035 |
Kampf C J, Jakob R, Hoffmann T. 2012. Identification and characterization of aging products in the glyoxal/ammonium sulfate system-implications for light-absorbing material in atmospheric aerosols[J]. Atmospheric Chemistry and Physics, 12(14): 6323-6333. DOI:10.5194/acp-12-6323-2012 |
Laskin A, Laskin J, Nizkorodov S A. 2015. Chemistry of atmospheric brown carbon[J]. Chemical Review, 115(10): 4335-4382. |
Li Y H, Yan Y L, Hu D M, et al. 2020. Source apportionment of atmospheric volatile aromatic compounds (BTEX) by stable carbon isotope analysis: A case study during heating period in Taiyuan, northern China[J]. Atmospheric Environment, 225(117369): 1-7. |
Lu Z F, Hao J M, Takekawa H, et al. 2009. Effect of high concentrations of inorganic seed aerosols on secondary organic aerosol formation in the m-xylene/NOx photooxidation system[J]. Atmospheric Environment, 43(4): 897-904. DOI:10.1016/j.atmosenv.2008.10.047 |
Maxut A, Nozière B, Fenet B, et al. 2015. Formation mechanism and yield of small imidazoles from reactions of glyoxal with NH4+ in water at neutral pH[J]. Physical Chemistry Chemical Physics, 17(31): 20416-20424. DOI:10.1039/C5CP03113C |
Moise T, Flores J M, Rudich Y. 2015. Optical properties of secondary organic aerosols and their changes by chemical processes[J]. Chemical Reviews, 115(10): 4400-4439. DOI:10.1021/cr5005259 |
Peng J F, Hu M, Du Z F, et al. 2017. Gasoline aromatics: a critical determinant of urban secondary organic aerosol formation[J]. Atmospheric Chemistry and Physics, 17(17): 10743-10752. DOI:10.5194/acp-17-10743-2017 |
Powelson M H, Espelien B M, Hawkins L N, et al. 2014. Brown carbon formation by aqueous-phase carbonyl compound reactions with amines and ammonium sulfate[J]. Environmental Science & Technology, 48(2): 985-993. |
Sato K, Takami A, Kato Y, et al. 2012. AMS and LC/MS analyses of SOA from the photooxidation of benzene and 1, 3, 5-trimethylbenzene in the presence of NOx: Effects of chemical structure on SOA aging[J]. Atmospheric Chemistry & Physics, 12(10): 4667-4682. |
Shao P Y, Tian H Z, Sun Y J, et al. 2018. Characterizing remarkable changes of severe haze events and chemical compositions in multi-size airborne particles (PM1, PM2.5 and PM10) from January 2013 to 2016-2017 winter in Beijing, China[J]. Atmospheric Environment, 189: 133-144. DOI:10.1016/j.atmosenv.2018.06.038 |
Shi X M, Wu F M, Jing B, et al. 2017. Hygroscopicity of internally mixed particles composed of (NH4)2SO4 and citric acid under pulsed RH change[J]. Chemosphere, 188: 532-540. DOI:10.1016/j.chemosphere.2017.09.024 |
Tajuelo M, Rodríguez A, Baeza-Romero M T, et al. 2019. Secondary organic aerosol formation from α-methylstyrene atmospheric degradation: Role of NOx level, relative humidity and inorganic seed aerosol[J]. Atmospheric Research, 230(104631): 1-10. |
Takekawa H, Minoura H, Yamazaki S. 2003. Temperature dependence of secondary organic aerosol formation by photo-oxidation of hydrocarbons[J]. Atmospheric Environment, 37(4): 3413-3424. |
Updyke K M, Nguyen T B, Nizkorodov S A. 2012. Formation of brown carbon via reactions of ammonia with secondary organic aerosols from biogenic and anthropogenic precursors[J]. Atmospheric Environment, 63: 22-31. DOI:10.1016/j.atmosenv.2012.09.012 |
Wang J, Zhang J S, Liu Z J, et al. 2017. Characterization of chemical compositions in size-segregated atmospheric particles during severe haze episodes in three mega-cities of China[J]. Atmospheric Research, 187: 138-146. DOI:10.1016/j.atmosres.2016.12.004 |
Wang H T, Ding J, Xu J, et al. 2019. Aerosols in an arid environment: The role of aerosol water content, particulate acidity, precursors, and relative humidity on secondary inorganic aerosols[J]. Science of the Total Environment, 646: 564-572. |
Wang L M, Wu R R, Xu C. 2013. Atmospheric oxidation mechanism of benzene. Fates of alkoxy radical intermediates and revised mechanism[J]. The Journal of Physical Chemistry A, 117(51): 14163-14168. |
Wang Y J, Luo H, Jia L, et al. 2016. Effect of particle water on ozone and secondary organic aerosol formation from benzene-NO2-NaCl irradiations[J]. Atmospheric Environment, 140: 386-394. |
徐俊, 黄明强, 冯状状, 等. 2018. 氨对甲苯二次有机气溶胶形成和化学组分的影响研究[J]. 环境科学学报, 38(8): 3243-3251. |
Yousefian F, Hassanvand M S, Nodehi R N, et al. 2020. The concentration of BTEX compounds and health risk assessment in municipal solid waste facilities and urban areas[J]. Environmental Research, 191(110068): 1-9. |
张威, 黄明强, 杨煜, 等. 2020. 酸性硫酸铵种子气溶胶对甲苯二次有机气溶胶形成和组分的影响研究[J]. 环境科学学报, 40(7): 2391-2399. |